 CMA nebo spinální svalová atrofie - je skupina několika typů genetických abnormalit, které vedou k částečné nebo úplné ztrátě motorických neuronů míchy v přední části.
CMA nebo spinální svalová atrofie - je skupina několika typů genetických abnormalit, které vedou k částečné nebo úplné ztrátě motorických neuronů míchy v přední části.
K tomuto problému dochází z důvodu mutace DNA SMN1 genu, který produkuje protein, - vzhledem k množství proteinu mutací v lidských pacientů pod normou, a to je zaručený ztráty motorických neuronů.
pro toto onemocnění se vyznačuje rozrušení příčně pruhovaných svalů nohou, hlavy a krku. Pacienti nemohou libovolně špatný tah:
- kraul;
- chodit;
- držet hlavu;
- vlaštovka.
ruce při práci jemné, citlivost a dobré duševní vývoj tam.
Content
- Historical
- O CMA přístupné a informativní
- Klasifikace typických rozdílů
- příznaky AMYOTROFIE Verdniga-Hoffmann
- nemoc Dubovica
- onemocnění Kyugelberga-Welanderova
- onemocnění v dospělosti
- Jiné druhy a typy SMA
- diferenciální analýzu
- problémy a ošetření
- Prevence
Historická fakta v roce 1891, CMA u dětí byla poprvé popsána vědec Verdniga. Ten jasně prezentovat popis patologických změn svalů, různé skupiny periferních nervů a míchy, přičemž poznamenal, atrofii buněk předních rohů míchy a kořenů.
V roce 1892, Hoffman řekl nosologických otázky nezávislosti.
V roce 1893 Verdniga a Hoffman poskytly důkazy o degeneraci míchy rohů buňky.
V roce 1956 byl předložen k veřejné diskusi později forma SMA, což má pozitivní výhled.
O CMA, které jsou přístupné a informativní
zajímavá fakta o spinální muskulární atrofie:
- navzdory vzácnosti tohoto problému, to je nejčastější porucha genetické formy;
- dětství onemocnění přenášené autosomnoretsessivnoy linie;
- gen mapována na chromozomu 5 a identifikovány v roce 1995;
- mezi dětmi průměrná porodnost se s tímto problémem 1. 6000 - 10000;1
- miminka formy muskulární atrofie nežijí až 2 roky v 50% případů;
- CMA může nastat nejen při narození, ale ve středním nebo stáří;
- průměrných hodnot každého z 50 osoby má recesivní gen;
- dítě, které má dva datové gen, a matky a otce na straně 25% náchylnější k porážce, pro normální zdravotní stav je postačující mít jeden normální gen.
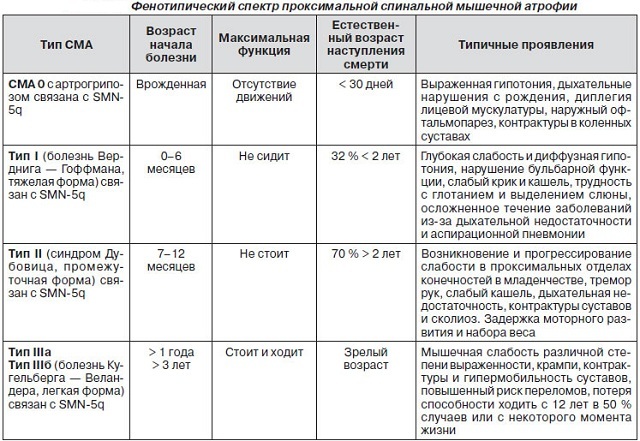
klasifikace a příznaky typické rozdíly
několik typů SMA.
AMYOTROFIE Verdniga-Hoffmann
píšu - dítě, Verdniga-Hoffmanova choroba, a to až na šest měsíců.Velmi těžký vzhled, tyto děti nemají sedět na vlastní pěst, hlava se nekoná, obtížné sání, polykání a dýchání, které se projevují fascikulace jazykové poruchy bulbární.
 nemoc je patrný již od prvních hodin po porodu, bez hlubokých reflexy a těžkou svalovou hypotonií.Výrazný osteoartikulární deformace. Dokonce i přidávání malého množství dětí, které budou moci sednout a držet hlavu tyto dovednosti brzy regresi.
nemoc je patrný již od prvních hodin po porodu, bez hlubokých reflexy a těžkou svalovou hypotonií.Výrazný osteoartikulární deformace. Dokonce i přidávání malého množství dětí, které budou moci sednout a držet hlavu tyto dovednosti brzy regresi.
Často tyto děti je vada kyčelního kloubu vývoje, koňskou nohou, přítomnost „kuře“ prsou.
počasí : onemocnění rychle postupuje, maligní formy. Smrt nastává až 9 let, z velké části kvůli problémům s kardiovaskulárním systémem a respirační selhání.Typ
onemocnění Dubovica
II - střední, Dubovica onemocnění, až na 18 měsíců.Tyto děti již mají za sebou první etapy pozitivního vývoje, ale nemohou naučit chodit, vývoj onemocnění je závislá na stupni patologie dýchacích svalů.
Obvykle symptomy spinální muskulární atrofie objevují v této podobě subakutní poté, co utrpěl důraz na fyzické zdraví, to začíná na dolních končetinách a postupně se přesune do horní části.
Objevuje se fascikulace jazyka, třes malých prstů, bulbulová paralýza.
Předpověď : mírnější maligní průběh, letální výsledek se objevuje ve věku 14 - 15 let. Typ
-Welanderova nemoc Kyugelberga
III - mládež, Kyugelberga-Welanderovu nemoc, od 18 do 30 měsíců.Tento typ je méně náchylné k smrti, dítě

snímku programátor Valery Spiridonov z Ruska - diagnóza spinální muskulární atrofie
sebeúcty, ale tento proces je pro něj velmi bolestivé, v budoucnu to bude neplatné, aniž by na invalidním vozíku nebo vycházkovou hůl se nepohybuje.
nemoc začíná potichu, se dítě začne chodit chůzi „mechanický panenku“, klopýtá a padá, pohyby stávají nejistá.Atrofie svalů je zpočátku špatně viditelná kvůli přítomnosti vyvinutého podkožního tuku.
Existují však všechny stejné příznaky třesu prstů, fascikulace jazyka, bulbárních poruch. Dokonce i v raných stádiích onemocnění zmizí dobře vyvinuté reflexe.
Klouby a kosti jsou deformovány, zejména v oblasti hrudníku.
Předpověď : děti ztrácejí schopnost chodit na 10 - 12 let a žijí až 20 - 30 let.
Nemoci v dospělosti
IV typ - dospělý, po 35 letech. Kvůli atrofii reflexů šlach, snížení proximálních svalů je člověk zbaven schopnosti samostatně se pohybovat.
Předpověď : kurz nemá vliv na očekávanou délku života.
Ostatní druhy a AGR
Dále spinální muskulární atrofie, přenášen na genetické úrovni, stále existuje určité množství souvisejících onemocnění:
- 1 typ X-vázaná amiatrofiya - start 40 - 60 let;
- 2 - vrozená, velmi agresivní forma, vede k smrti až 3 měsíce;
- 3 - jsou postiženy všechny končetiny, téměř ve všech případech trpí chlapci;
- distální spinální amyotrofie typu 1 - od narození, někdy in utero, jsou postiženy horní končetiny;
- 2 a 4 typy - toto onemocnění bylo popsáno pouze v jedné rodině;
- 3 je pomalu se rozvíjející onemocnění;
- 5 typické - pomalu se rozvíjející v mladém věku;
- typu VA a VB - porážka horní končetiny;
- porážka holeně - objevuje se v dospívání, rozvíjí se pomalu se slabostí kolen;
- léze hlasivky - paralýza vazů u dospělých;Autozomální dominantní spinální amyotrofie
- - objevuje se v dospělosti a její mladistvá forma v dospívání;
- je vrozený charakter - onemocnění z prvních okamžiků života;
- skapulární fibulární druhy - velmi vzácné onemocnění spodních končetin s zakřivením chodidla;
- juvenilní segmentový typ lézí míchy - velmi vzácných případech, které se projevují v mládí s pokrokem 2 - 4 roky po postiženém rameni;
- Fenkelova nemoc - se rozvíjí hlavně kolem 37 let, existují však případy výskytu ve věku 12 let;
- Jockelova choroba je pozdní a pomalý nástup;
- spinální amyatrofie s myoklonickou epilepsií - jsou postiženy končetiny, většinou kvůli myoklonickým záchvatům;
- páteřní forma s vrozenými zlomeninami kostí - těžká forma svalového vyčerpání;
- pontocerebelární hypoplazie - existuje 8 typů;Asymetrická segmentová forma
- - onemocnění 15 - 25 let starých lidí.

Diferenciální analýza
Předpověď je možná z důvodu získaných dat vyplývajících z: genetické analýzy
- ;
- charakteristiky kliniky( typ onemocnění, umístění lokalizace);Příznaky asociované s
- , například fascikulace jazyka;Výsledky
- údajů o EKG a biopsii kostních svalů.
Rozlišovat kojence a časná forma SMA může být založeno na problémech s vrozenou hypotonií - syndrom „poddajný“ dítě amiatonii, vrozené svalové dystrofie benigní povahy, dědičnost a analýzu chromozomů.druh
mládeže se liší od páteře amiatrofii Kugelberg-Veelander a různé typy svalové dystrofie.
úkoly a procedury
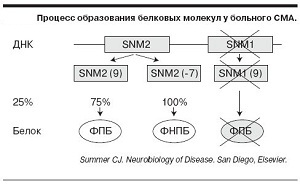 Vzhledem k tomu, nemoc se vyskytuje především v důsledku nedostatku bílkovin SMN- je hlavním cílem léčby je zvýšit svou úroveň.Dnes v tomto směru aktivní výzkumné ústavy v USA, Itálii a Německu.
Vzhledem k tomu, nemoc se vyskytuje především v důsledku nedostatku bílkovin SMN- je hlavním cílem léčby je zvýšit svou úroveň.Dnes v tomto směru aktivní výzkumné ústavy v USA, Itálii a Německu.
Díky skupiny dobrovolníků k provádění klinických zkoušek terapie SMA s kyselinou valproovou, butyral sodný a jiné léky.Údaje o využití kmenových buněk dosud zveřejněny.
na stavu pacientů má pozitivní efekt:
- masáž;

- neuromuskulární stimulace;
- fyzioterapie, jakékoliv fyzické aktivity jsou jen pozitivní účinky. Pacienti
- SMA potřebují dietu;podávání
- ESP léků, které obsahují vitamin komplexy, léky proti bolesti, zlepšení metabolismu nervové tkáně a svalů, jejich stimulace.
Dnes je stav pacienta může být udržována a trochu jednodušší, ale spinální muskulární atrofie je nevyléčitelná.
Prevence Preventivní opatření spočívají v pasivních činnosti - poradenství pro rodiče, kteří mají gen CMA o možném riziku, prenatální diagnostiky DNA před 14. týdnem těhotenství, klků biopsie choriových pro konečné diagnóze a rozhodování o dalším postupu rozhodnutí rodičů.