 olivopontocerebelární degenerace( OPTSD) as hotelového typu onemocnění byla popsána již před více než 100 lety. Po celou dobu, kdy byla choroba studována, byly vyvinuty metody diagnostiky a léčby.
olivopontocerebelární degenerace( OPTSD) as hotelového typu onemocnění byla popsána již před více než 100 lety. Po celou dobu, kdy byla choroba studována, byly vyvinuty metody diagnostiky a léčby.
Dnes je známo, že olivopontocerebelární degenerace je zděděna. Onemocnění postihuje centrální nervový systém. Lokalizace atrofie - cerebellum, můstek a dolní olivy( části mozku).
Hlavním projevem je změna v obvyklém chování - dochází k ataxii, extrapyramidovým poruchám. Tento typ onemocnění se vyznačuje délkou trvání kurzu, ale je fixován v dospělosti. V procesu změn v lidském mozku, označený smrti nervových buněk, v daném pořadí porušil frekvence signálů z nich v CNS.
Také degenerativní-dystrofické procesy, které se vyskytují po celou dobu vývoje onemocnění, postihují mozkové kůry, můstek a olivový mozek.
To je důvod, proč existují charakteristické změny v psychice a možnosti člověka, včetně fyzické.Ve vzácných případech, během diagnózy 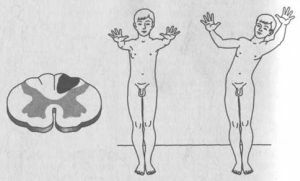 , jsou léze zaznamenány v oblastech, jako je přední roh míchy a její cesty. Onemocnění může také ovlivňovat kaudální kraniální nervy( 9-12 párů).Postihy spojené s olivopontocerebelární degenerací zahrnují: Alzheimerovu chorobu
, jsou léze zaznamenány v oblastech, jako je přední roh míchy a její cesty. Onemocnění může také ovlivňovat kaudální kraniální nervy( 9-12 párů).Postihy spojené s olivopontocerebelární degenerací zahrnují: Alzheimerovu chorobu
- ;
- roztroušená skleróza;
- Pickova nemoc.
Dědičnost onemocnění nastává na recesivní a dominantní cestě a jsou známy i případy sporadického dědičnosti.stav
pacienta může být vyjádřen ve ztrátu orientace, paměťové výpadky, úplné nebo částečné skleróza, poruchy chůze, tuhost, snížení motorické aktivity obecně, a tón tělo, letargie, ztráta schopnosti soustředit.

Statistické údaje
Většina případů onemocnění je hlášena u starších osob - po 60-65 letech - 75% případů.
však degenerace může začít rozvíjet v raném věku - po dobu 30-40 let a v případě dědictví a u dětí ve věku 10-11 let( 2-3%).
To je důvod, proč musíte být vysílán nejen pro dosažení starších lidí, ale v 20-40 letech, a v případě vrozených vad - průběžně po celý život.
příčin nemocí
Důležitým rysem onemocnění je fakt, že přesné příčiny jejího vzniku a vývoje, pokud žádný případ onemocnění v rodině, žádný známý lék ani sto let po prvním popisu. V místech genů dochází k porušování, což se projevuje zvýšením počtu trinukleotidových opakování.
Mezi další možné příčiny, které mohou vést k výskytu charakteristických poruch, jsou zaznamenány asymetrické atrofické změny v bílé hmotě.Atrofie cerebrální kůry je poznamenána v pozdních stádiích onemocnění.Abychom zjistili přesné důvody, je nutné důkladně prošetřit.
Odrůdy proudit
Existuje několik základních typů olivopontocerebelární degenerace: Typ 
- Mendela;
- typ Finkler-Winkler;
- atrofuje degenerací sítnice;Typ
- Shut-Haikman;
- atrofuje s demencí.
také samostatná kategorie může zahrnovat Shay syndrom - Dreydzhera, která je charakterizována následujícím základní frekvenci příznaků:
- autonomní dysfunkce;Cerebrální poruchy
- a ataxie( různé stupně závažnosti);
- léze bazálních ganglií.
Type Mendel má autosomálně dominantní dědičnost a mechanismus z následujících příznaků:
- pomalý a nudný vývoj onemocnění, který vyvolává jeho vývoj a přechod na zanedbanou fázi;
- věk manifestace prvních symptomů - od dětství do 60 let;Ataxie
- ;
- snižování svalového tonusu( někdy významné, člověk nemůže provádět rutinní úkoly);Poruchy řeči
- ( projevily se ze slabé na silné);
- třes a třes v rukou( třes);Porucha
- při polykání;Hyperkineze
- ;Poruchy
- spojené s pohybem očí( zřídka).
Degenerace Flicler-Winkler se projevuje následujícími vlastnostmi:
- je dědičná autosomálně recesivní metodou;
- věk nástupu příznaků a vývoj nemoci v průběhu života, počínaje věkem 20 let;
- ataxie končetin.
Pokud je tento druh diagnostikován, neexistují žádné důkazy o narušení motorické aktivity.
Typ s degenerací sítnice má následující příznaky: 
- je autosomálně dominantním přenosovým principem pro novou generaci;
- postihuje mladé lidi a děti;Ataxie
- ;Extrapyramidové poruchy
- ;
- změny zrakové ostrosti( významný pokles), dochází k porušení v důsledku pigmentace sítnice.
Vzhled Jester-Haikman je charakterizován následujícími příznaky a projevy:
- je autosomální dominantou - hlavní metodou dědičnosti;
- projevy a vývoj mladých( 20-30 let) nebo dětství;Ataxie
- ;
- charakteristická pro tento typ symptomů - obličejová paralýza;
- poruchy polykání( někdy závažné);Poruchy řeči
- ;
- porucha vibrací.
Typ s demencí vyznačující se následujícími vlastnostmi: autozomální dominantní dědičnost
- ;
- projev a vývoj - ve středním věku, ale nejpozději do 40 let;
- porušení inteligence( někdy velmi silné);Extrapyramidové poruchy
- ;Ataxie
- .
Příznaky jsou obecně podobné, ale současné rozdíly umožňují lékařům správně stanovit diagnózu a předepisovat nejúčinnější léčbu.
Společné příznaky
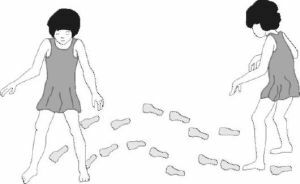
Společným příznakem pro všechny typy olivopontocerebelární degenerace je ataxie. Na počátku se u člověka objevuje mírná nestabilita, pak při rychlém chůzi dochází k nepohodlí pacienta, projevující se v nevhodných pohybech. Další vizuální poruchy chůze: časté pádu
- ;
- široce od sebe při chůzi pěšky;
- fluktuace těla ze strany na stranu( narušení cerebellum);
- nestabilní pozice při sedění na židli.
Pozdní symptomy( průběh onemocnění): velký rukopis 
- ;Dysmetrie
- ;
- se třes v končetinách( třes);
- chvění hlavy;
- snížil nebo zvýšil reflexy;Poruchy řeči
- .
Mezi obvyklé příznaky patří: porucha polykání
- ( ve většině případů je zaznamenána);Pyramidální selhání
- .
 Obličejová paréza není běžným příznakem pro všechny typy, na rozdíl od inkontinence.
Obličejová paréza není běžným příznakem pro všechny typy, na rozdíl od inkontinence.
Člověk se téměř vždy stává bezdomovcový a apaticky, ztrácí zájem o události kolem sebe.Často je to opožděné zpomalení a dokonce i hloupost. Také onemocnění je doprovázeno demencí v různých stupních závažnosti, depresivními stavy, strachy a fóbií, které se dříve objevily. Halucinace a zmatenost jsou charakteristickými příznaky této nemoci.
Pokud je nemoc je ve stavu zanedbávání, člověk postupně ztrácí schopnost chodit na první pohled, a pak přestane vykonávat jednoduché kroky k péči o sebe.Často během tohoto období pacienti oslabují imunitu, což vede k rozvoji souběžných onemocnění.Diagnostické kritéria
Abychom diagnostikovali onemocnění a pak přesně věděli její typ, bude trvat několik vyšetření.To je nezbytné pro získání objektivních a spolehlivých informací.Jedním z vyšetření je neurologický stav. Vyšetřovány jsou také neuropsychologické údaje.
Všechny výsledky umožňují lékaři zjistit, zda pacient má degeneraci cerebellar, zda je přítomna roztroušená skleróza a další příznaky olivopontocerebelární degenerace.
Objektivní informace lze získat tím, že prochází počítačovým tomografií skenování mozku a poté MRT mozku. K určení druhu onemocnění budete potřebovat poradit se specialistou v oblasti genetiky.
Dále může být vyžadována diagnostika DNA.Pokud dojde ke značnému zhoršení vidění, diagnostika se přidá ke konzultaci s oftalmologem.
Komplex opatření - co lze udělat?
Nebyla vytvořena žádná zvláštní účinná léčba této choroby. V současné době se provádějí opatření k udržení ukazatelů v průměru a blíží se k normálním hodnotám. Ve většině případů se provádí symptomatická léčba s předpojatostí v neurologii.
Rovněž zdůrazňuje symptomatologii přítomnou v době diagnózy.
Základní léky - neurometabolity, anticholinergika a také posilující látky. Pro zachování optimálního svalového tonusu jsou poskytovány terapeutické masáže a cvičební terapie. Takové léky se také používají: kyselina glutamová 
- ;
- Cerebrolysin( injekce);
- Aminalon;
- Proserin.
Aktivní venkovní aktivity a jednoduché procházky jsou součástí komplexu opatření pro léčbu onemocnění.Pokud jsou zrakové postižení, pak terapie zahrnuje vhodné léky.
významná zlepšení lidského zdraví, nebudou hodnoceny. Nicméně strávil terapie bude udržovat výkon v přijatelných po dlouhou dobu, takže doba trvání nemoci v průměru 12 let, existují případy, kdy pacienti mohli bojovat takového onemocnění po dobu více než 20 let. Hlavní příčinou úmrtí - zápal plic nebo sepse jako komorbidit.