 degenerativní-dystrofické onemocnění nervového systému s převažující zapojením periferních nervů a svalových vláken, zaujímají velký podíl ve struktuře lidských dědičných onemocnění.
degenerativní-dystrofické onemocnění nervového systému s převažující zapojením periferních nervů a svalových vláken, zaujímají velký podíl ve struktuře lidských dědičných onemocnění.
Typickým zástupcem je myotonická dystrofie( nebo dystrofické myotonie) popsal na začátku minulého století, několik autorů a nazval nemocí Rossolimo-STEINERT-Kurshmana.
Tato nemoc je nejznámější chorobou v kategorii myotonie a nejčastější formou svalové dystrofie u dospělých. Co je to tato nemoc a jak ji bojovat?
Otevření a jsou
Rossolimo onemocnění, Steinert a Kurshmana studoval choroby jsou genetické poruchy s autosomálně dominantní dědičnosti. To znamená, že jeden rodič má mutantní gen, nemocné děti se rodí s pravděpodobností 50%.Nemoc má povahu rodinné úzkosti a je přenášena na následující generace ve vertikálním měřítku.
Synové a dcery v těchto rodinách jsou nemocné se stejnou frekvencí, přibližně 3 až 5 osob na 100 tisíc obyvatel. Věk nástupu onemocnění, stejně jako závažnost symptomů, jsou výrazně variabilní.
popsat časné neonatální a pozdní formy, ale často se nemoc debutuje na druhém, přinejmenším - ve třetí dekádě života.  Je třeba poznamenat, že přenos nemoci z matky na dítě je prognosticky nepříznivější než u otce.
Je třeba poznamenat, že přenos nemoci z matky na dítě je prognosticky nepříznivější než u otce.
Základem onemocnění gen vady je 19 párů chromozomů, které je zodpovědné za syntézu enzymu proteinové kinázy-miotonin. Tento protein je obvykle přítomen nejen v kosterním svalstvu, ale také v buňkách myokardu a centrální nervové soustavy.
Proto jsme pro dystrofickou myotonii charakterizovali polysystémové projevy s porážkou různých orgánů a systémů.Méněcennosti miotonin kinázy vede k svalové křeče, spolu se změnami v atrofické svalstva hlavy, krční páteře, končetin. Tam
kombinace hypertrofii svalových vláken s jiným atrofií a jejich nahrazení v tuku nebo pojivové tkáně.
Klinické projevy
Vzhledem k variantě nástupem onemocnění v klinické praxi jsou tyto formy v závislosti na věku:
- kongenitální formy - projevem onemocnění začíná bezprostředně po narození dítěte;
- mládežnická verze - debut myotonie ve věku od jednoho roku do puberty;
- klasická forma - začátek klinických projevů spadá do druhého a třetího desítky životů;
- je minimální verze - projev spadá na pozdní termíny - šesté desítky života.
Zdá se, že čím později se onemocnění projevuje, tím příznivější je průběh a tím lepší prognóza. Mezi nejčastější klasická forma STEINERT choroby, pro které tyto klinické příznaky jsou typické:
- myotonií - projevuje křečemi žvýkacích svalů a flexor svaly na rukou, vyznačující se tím, atrofické změny v různých
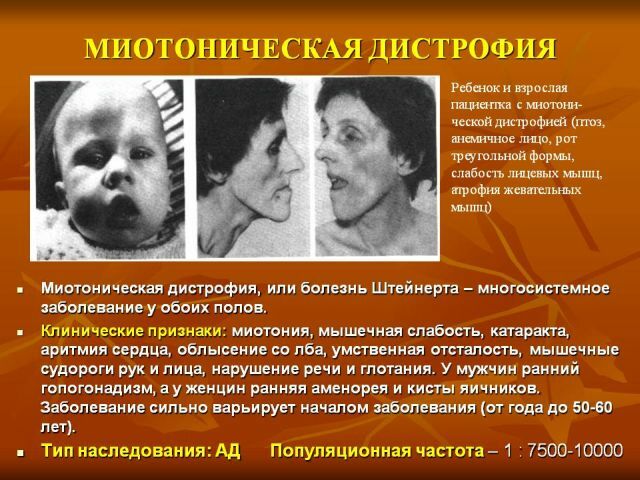
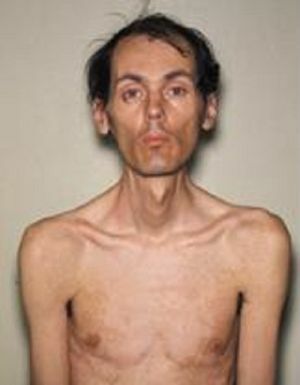 svalových skupin. Postupně blednutí myotonická symptomy a progresi svalové dystrofie, z vnějšku je vyjádřena v smutný obličejové masky a obličejové nepřítomnosti. Nebezpečný je paréza svalů hrtanu s poruchou polykání, a slabosti dýchacích svalů, což vede k možným útokům apnoe během spánku, vývoje pneumonie.
svalových skupin. Postupně blednutí myotonická symptomy a progresi svalové dystrofie, z vnějšku je vyjádřena v smutný obličejové masky a obličejové nepřítomnosti. Nebezpečný je paréza svalů hrtanu s poruchou polykání, a slabosti dýchacích svalů, což vede k možným útokům apnoe během spánku, vývoje pneumonie. - Kardiovaskulární poruchy - srdeční arytmie, hypertrofické změny z levé komory, zjištěné na elektrokardiogramu, městnavého srdečního selhání.
- Endokrinní poruchy( hlavně ovlivněna sexuální funkce) - snížení velikosti pohlavních orgánů, snížení sexuální touhy u žen - poruchy menstruace, obezita.
- General mění dystrofický - suchost a pigmentaci pokožky, ztrátu části nebo všechny vlasy a zuby, časně šedý zákal.
- Poruchy CNS - únava, poruchy spánku, apatie, ztráta inteligence.
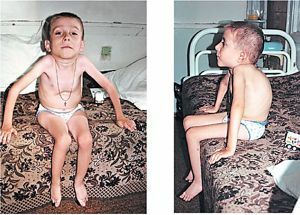
Samostatně stojí za zmínku, typické klinické projevy vrozených forem dystrofickým myopatie:
- snížení aktivních fetálních pohybů v děloze, zjištěných během ultrazvuku;
- v novorozeneckém období - letargie, hypotenze rozšířené, a to zejména v žvýkání, pleťové, svaly bulvy;
- zachování a dokonce zvýšení reflexů šlach;
- problémy s podáváním, respirační potíže jako syndrom respirační tísně;
- zpoždění fyzického a neuropsychického vývoje, příznaky oligofrenie;
- rychlá progrese onemocnění, vysoké riziko náhlé smrti. Kritéria
Diagnostické
podezření na nákazu Rossolimo-STEINERT - Kurshmana může dojít v lékařem, pokud má pacient kombinaci myotonická dystrofie a změny ve svalech na pozadí ztráty inteligence a přítomnost kardiovaskulárních a endokrinní onemocnění.
Polysystemic téměř vždy indikuje genetickou povahu onemocnění.Tito pacienti jsou předmětem analýzy DNA a vedení genealogický analýzu k potvrzení dědičnost autosomálně dominantní onemocnění.Jako informační výzkumné metody používané elektrokardiografie, electroneuromyography, analýzy hormonů.
Vzhledem k univerzálnosti klinických projevů procesu stanovení diagnózy jsou většinou jednalo o odborníky z různých lékařských oborů - genetiky, kardiologie, endokrinologie, gynekologie, andrologie, neurologie.
Diferenciální diagnóza mezi dystrofické myotonií a jiné typy podobných onemocnění.Na rozdíl od ostatních je svalová atrofie charakteristická pro Rossolimovou chorobu.Často k potvrzení diagnózy je nezbytné uchýlit se k biopsie k určení úrovně svalové bílkoviny, které v tkáních v této patologii zvýšené.
Antenatální diagnostika se provádí také metodou výzkumu plodové tekutiny.
Lékařská pomoc
genetické onemocnění, nelze vyléčit úplně, takže cílem léčby u onemocnění Rossolimo-STEINERT-Kurshmana je příznak úlevy, zlepšení celkového stavu a sociální adaptaci pacientů.principy
léčby jsou následující: soli
- strava s nízkým draslíku( jablka, chřest, zelí, okurky, vinná réva, zelenina, kukuřice, jahody, ředkvičky, mandarinky, grapefruity
 , cibule, mrkve, lilek, hrachu);
, cibule, mrkve, lilek, hrachu); - potlačení podchlazení , aby se zabránilo křečemi;Použití
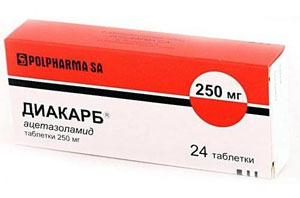
- léků chinin ke stabilizaci buněčné membrány těchto léčiv, jako Difenin, prokainamid, Diakarb - ke zmírnění svalové křeče a nižší tuhosti, záchvaty, snížení intrakraniálního tlaku;
- užívání anabolických steroidů ( Metanandrostenolon, Retabolil, Nerabol), vitamíny, ATP stimulovat svalovou hmotu;
- pohybová terapie, masážní, elektrické, ortopedické zařízení.
Tyto události poskytují dobrou pozitivní vliv jak v klasické, tak i ve formě vrozené onemocnění.Zcela zbavit pacienta, ze kterého nemohou dystrofní myotonií, ale prodloužit život a zlepšit jeho kvalitu plechovku.
horší prognóza vrozených forem - úmrtnost je vysoká, děti nemohou žít až 3 roky. Junior verze myotonií dochází 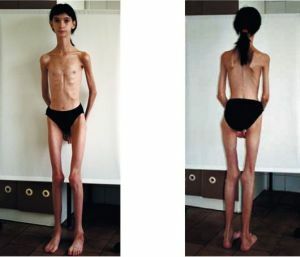 poměrně obtížné a může vést již v mladém věku k omezení pracovní neschopnost a předčasný postižení.
poměrně obtížné a může vést již v mladém věku k omezení pracovní neschopnost a předčasný postižení.
V případě klasické formy onemocnění může dojít dlouhou dobu při provádění včasné léčby a rehabilitačních činností.Nejpříznivější prognóza v pozdních onemocněních onemocnění.
Preventivní opatření jsou omezeny na to, že ženy ze znevýhodněných rodin s historií v plánovací fázi těhotenství je nutné být testovány na přítomnost abnormálních genů zodpovědných za rozvoj svalové dystrofie. Je také vhodné, pokud je přítomna patologická situace otcovy příbuzné.
příležitosti narození dětí se musí řešit individuálně v každém jednotlivém případě, že lékař - genetika po konzultaci.