Angelman Syndrom( "happy marionet" syndromet, "Persille Syndrome") - er en sjælden neurologisk lidelse relateret til kromosomafvigelser.
Angelman Syndrom( "happy marionet" syndromet, "Persille Syndrome") - er en sjælden neurologisk lidelse relateret til kromosomafvigelser.
Ca. tusind nyfødte har et barn med denne patologi. Hos patienter, der oplever fysiske og intellektuelle forsinket udvikling, søvnforstyrrelser, kramper, krampeanfald, rykvise bevægelser, privat årsagsløse latter.
Børn med denne patologi ser altid meget lykkelige ud.
Content
- historie undersøgelse
- sygdom patogenese og forårsager
- genetiske mutationer øger risikoen for
- gruppe af symptomer på sygdommen
- kliniske billede i detaljer
- diagnose og rettidig identifikation
- Foto og videomateriale om emnet
- Forbedring af tilstanden og livet af patienterne
- Funktioner af uddannelse og tilpasning
Historiestuderer sygdomme
sygdommen er opkaldt efter Harry Angelman, en britisk børnelæge, der først differentieret syndromet i 1965. Derefterog han blev udnævnt en lykkelig marionet syndrom, men udtrykket bruges ikke i dag, da det blev anset for nedsættende.
Dr. Angelman været at behandle flere børn med lignende symptomer og foreslog tilstedeværelsen af en fælles diagnose. Bevis diagnosen, og for at få præcise data på den tid, var det ikke muligt på grund af mangel på teknologier, der er til rådighed i dag. Hans teorier lægen afspejles i en artikel kaldet "Children marionetter."
Mens publikationen ikke har skabt stor interesse for offentligheden, og blev hurtigt glemt. Endnu engang blev det husket i firserne, da de nødvendige forskningsmetoder kom til udtryk. Forskere har fundet, at en stor andel af børn, der lider syndromet, er der ingen lille del af de 15 kromosomer.
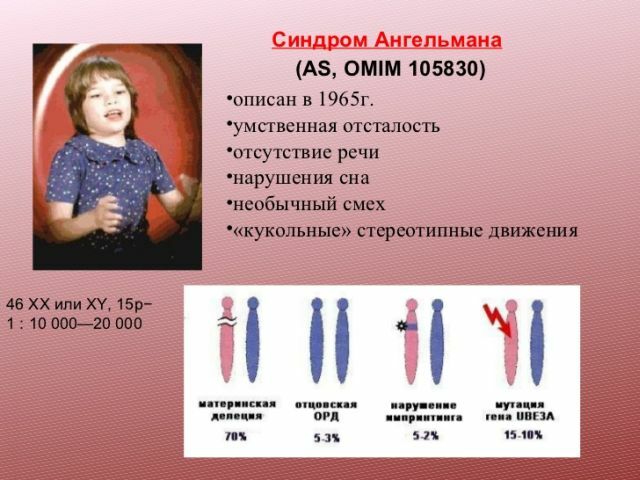
Patogenese og deres årsag
Denne defekt opstår på grund af deletion( fase tab af genetisk materiale) segment af kromosom 15.Blandt andre årsager, ofte kaldet odnoottsovskuyu Dis( fædrenearven af to kopier af kromosom stedet for kopier fra begge forældre), translokation eller mutation af et enkelt gen.
syndrom kan også forekomme som et resultat af en genmutation involveret i metabolismen af ubiquitin.
fleste patienter med denne patologi i familiens historie er ingen genetiske abnormiteter. For en vis procentdel af patienter er dette syndrom imidlertid arvet. Risici er højere, hvis forældrene har kromosomale abnormiteter. Interessant faktum - hvis familien havde et barn med dette syndrom, så er sandsynligheden for fødslen af et barn med den samme patologi, er 1%.
udseende Angelman syndrom ganske spontant, og næsten hver familie kan have et barn med denne sygdom. Der er ingen specifikke statistiske data om antallet af børn med dette syndrom.
Ifølge forskellige skøn af antallet af spædbørn med syndromet dukker fra ét til ti tusind til en ud af tyve tusind. Forskere foreslår, at dette nummer faktisk er meget større.
genetiske mutationer øger risikoen for fremkomsten af sygdomme
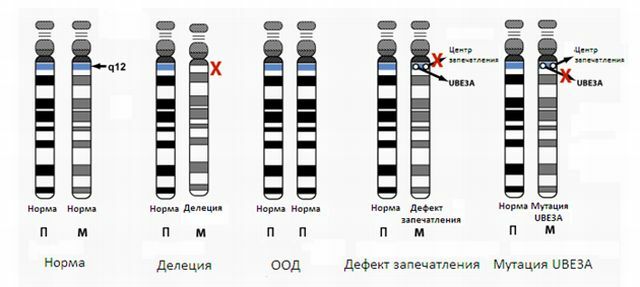
Angelman syndrom er forbundet med tilstedeværelsen af forældrene til barnet forskellige kromosomafvigelser. Blandt sådanne afvigelser kaldes normalt:
- trisomi af kromosom - tilstedeværelsen af en eller flere ekstra kromosomer i kromosomet sæt;
- inversion - tilbageførsel af en af de kromosomer dele 180 grader, den del af kromosomet er udeladt, og generne er anbragt i den modsatte rækkefølge;
- mikrodeletioner , som er resultatet af justering og Y-kromosom-segmenter mellem kromosomer udveksling, der er et lille antal kromosomer, og der kan være nogen af generne;
- sletning af - mangel på en af kromosomsektionerne;
- translokation af - overførsel eller vedhæftning af en region af et kromosom til et andet kromosom;
- dobbeltarbejde af - kopiering af del af kromosomerne, hvilket resulterer i overskydende genetisk materiale;
- ringformet kromosom - på kromosom ender offline genetisk materiale, er de nydannede ender forbundet i en ring.
Genmutationer, der kan forårsage syndromet udvikler symptomer
Grupper Normalt diagnosen Angelman Syndrom er sat i alderen 3 og 7 år, når symptomerne bliver udtalt. Nyfødt baby syndrom adskiller sig ikke fra raske børn, men i løbet af de første måneder af livet, der er problemer med fodring, børn langsomt tager på i vægt, har problemer med at sove, forsinkelse i motorisk udvikling.
Alle symptomer på persille syndrom er opdelt i flere typer. De har et forhold til neuralgic, mentale og fysiske tilstand af patienten:
- De fysiske symptomer indbefatter problemer med synet( skelen, optisk atrofi), udragende tunge, mund bred,
 stor afstand mellem tænderne, hovedstørrelse proportionalt mindre i forhold til andre dele af kroppen størrelse, albinisme og hypopigmentering, inflexion af fødderne af fødderne, når de går( altså sammenligningen med marionetter), skoliose.
stor afstand mellem tænderne, hovedstørrelse proportionalt mindre i forhold til andre dele af kroppen størrelse, albinisme og hypopigmentering, inflexion af fødderne af fødderne, når de går( altså sammenligningen med marionetter), skoliose. - observere rysten i lemmerne, en bred vifte af søvnforstyrrelser, hysteriske anfald, hvis intensitet er høj i en alder af tre år, og efterhånden aftager, når de bliver ældre, de problemer i processen for kommunikation( tale udvikling forsinkelse), hyperaktivitet og opmærksomhed underskud Blandt neurologiske symptomer.
- Psykologiske symptomer er normalt udtrykt i en alt for emotionel adfærd( konstant glad udseende, venlighed), mental retardering, alvorlig sygdom.
Klinisk billede i detaljer
Forskellige patienter kan udvise forskellige symptomer. Denne forskel afhænger af graden og typen af kromosomal abnormitet.
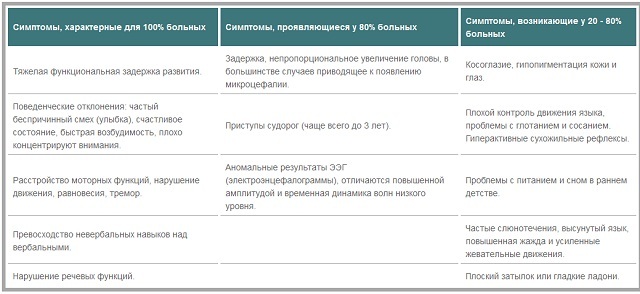
kan opdele symptomerne med hyppigheden af deres udseende i børn med Angelman syndrom:
- symptomer, der opstår hos alle patienter .Disse omfatter svær funktionel forsinket udvikling, adfærdsmæssige problemer( griner og smiler, uden grund, at tilstand af lykke, irritabilitet, nedsat koncentration af opmærksomhed).Også i absolut alle patienter er der en krænkelse af motorfunktioner, ubalance, tremor, overvejelsen af ikke-verbale færdigheder over verbale, taleforstyrrelser.
- Symptomer typisk for 80% af patienter. Forsinkelse i vækst, hvilket fører til en disproportion af hovedet i forhold til andre dele af kroppen. Dette fører ofte til udviklingen af mikrocefalier. Hos børn under tre år opstår epileptiske anfald ofte, så bliver de mindre hyppige eller forsvinder helt. Resultaterne af elektroencefalogrammet er ofte uregelmæssige, bølger med lavt niveau har en øget amplitude og temporal dynamik.
- Symptomer der opstår hos mindre end 80% af patienter. Disse manifestationer omfatter skelen, nedsat evne til at styre bevægelser af tungen, problemer med at synke, hypopigmentering af hud og øjne, albinisme, der mange patienter er øget aktivitet af senereflekser. I tidlig barndom har mange problemer med ernæring og søvn. Blandt manifestationerne kan også kaldes salivation, poking ud af tungen, konstant tørst. Eksterne funktioner omfatter tilstedeværelsen af en flad nakke og glatte palmer.
Når du vokser op, ændrer manifestationerne af syndromet. Søvnforstyrrelser, hyperaktivitet, konvulsiv syndrom er mindre almindelige. Voksne, der har Angelmann syndrom, ser yngre ud end deres jævnaldrende.
Puberteten sker lidt senere end hos personer uden sygdommen. Patienter kan have deres egne børn, men i høj risiko for at overføre sygdommen til afkommet.
Mange voksne patienter lider af ukontrolleret vandladning. Ofte fandt eksistensen af vanskeligheder med motorikken, hvilket resulterer i et behov for at bære tøj uden lynlåse, knapper. Blandt voksne patienter har problemet med overvægt, så det er nødvendigt at overvåge overholdelsen af en speciel diæt.
Børn er modtagelige for det talte ord, og forstå indholdet i næsten alle samtaler, men sjældent mødes. De har ofte nægter at deltage i samtalen, og brug i en tale et par dusin ord.
diagnose og rettidig identifikation
syndrom Doll muligt at diagnosticere før fødslen, gennem genetisk forskning femtende kromosom.
Diagnose kan være invasiv og ikke-invasiv. Invasive studier i medicin der er lang tid, men er snarere risikabelt, som du har brug for at trænge ind i livmoderen og fosterhinde væskeindtag.
ikke-invasiv metode involverer analyse af moderens blod, hvor barnet udføres DNA-analyse. Baseret på denne undersøgelse er der truffet konklusioner om tilstedeværelsen eller fraværet af visse abnormiteter.
Diagnostics også holde en nyfødt, der observerede en overtrædelse af muskeltonus, forsinket udvikling af tale og motoriske færdigheder.
er nødvendigt at overvåge barnet ansigt, hans opførsel, en manifestation af følelser, bevægelser. Hvis barnet har svært med at bøje benene, er der en rysten, kaotisk og pludselige bevægelser af lemmerne, bør du søge råd og bistand fra fagfolk.
Ofte er diagnosen Angelman Syndrom sat i alderen 3 og 7 år, når du vil bemærke en levende manifestation af symptomer på sygdommen. Afhængig af graden af beskadigelse af kromosom 15, kan manifestationer være af forskellig sværhedsgrad: nogle patienter har svært ved selv i tale, mens andre kan føre et uafhængigt liv.
Foto og videomateriale om emnet
Video, samt et galleri med billeder af børn og voksne med diagnosen Angelman syndrom:





forbedre tilstanden og livet af patienterne
Angelman syndrom er en genetisk sygdom, langtdag er der ingen recovery metoder til kromosomafvigelser, og behandling er ikke mulig. Men der er mange måder at reducere symptomerne, hvilket gør det nemmere at angive disse mennesker.
I hvert tilfælde er det rehabiliteringsprogram individuelt designede henholdsvis og symptomerne på en bestemt patient. Eksperter identificere fire hovedområder behandling:
- Modtagelse antiepileptika og antikonvulsiva .Disse stoffer hjælper med at kontrollere og reducere hyppigheden af angreb
 forårsagede sygdom.
forårsagede sygdom. - Terapeutisk motion - hjælper med at udvikle finmotorik og løse andre problemer bevægeapparatet. Børn med abnorme kromosomer udvikler langsommere end deres sunde jævnaldrende, og det kræver tålmodighed på den del af familien.
- tegnsprog .Patienter med marionet syndrom taler lidt, men bruger ganske rigtigt tegnsprog. Træningen skal starte fra en meget tidlig alder.
- Adfærdsterapi .Dette program giver os mulighed for at give en korrekt og effektiv undervisning af børn, der har abnormiteter til at hjælpe med hyperaktivitet og opmærksomhed underskud.
Mange læger noterer ligheden mellem børn med autisme og Angelman syndrom. Amerikanske forskere har gjort fremskridt i behandlingen, nemlig brugen af intravenøse injektioner med hormonet Secretin. Den positive effekt udtrykkes i at reducere tegn på uønsket adfærd, samt at forbedre kommunikative færdigheder.
Uddannelses- og tilpasningsfunktioner
Da barnet vokser op med denne patologi, ændres symptomerne, bliver mindre intense, eller tværtimod mere intense, de gamle kan forsvinde, og nye kan forekomme. Udsigter i patientens tilstand afhænger i vid udstrækning af de forhold, der omgiver patienten.
Et velvilligt miljø giver atmosfæren af kærlighedssammenhæng patienten chancen for at blive, i det mindste delvist, en uafhængig person. Med passende pleje bliver tegn på sygdommen lettere med tiden. Forventet levetid hos personer med Anghelman syndrom er gennemsnitlig. Det er nødvendigt at huske om regelmæssige undersøgelser, korrekt ernæring samt overholdelse af betingelserne for terapi.
Undervisning af et barn i skolen kræver brug af inkluderende programmer. Men i Rusland og CIS-landene i dag er disse programmer ikke omfattet af en bred praksis. Sådanne børn har brug for en særlig tilgang, som omfatter uddannelse i koncentrationen af opmærksomhed. Et vigtigt krav er også muligheden for, at uddannelsesinstitutionen yder lægehjælp i tilfælde af pasform.
I vores samfund er der for øjeblikket personer med kromosomale abnormiteter forsigtige og forsigtige. I de senere år har forskellige aktiviteter været aktivt udført med det formål at sprede viden om sådanne sygdomme samt skabe tolerance i samfundet.