Nykyään on olemassa useita orpo( harvinainen) sairaudet, jotka eivät pysty käsittelemään nykylääketieteen. Mahdollisuus on olemassa vain tukea näitä potilaita osittain parantamalla heidän elämänlaatuaan, mutta voittaa sairaus on mahdotonta.
Nykyään on olemassa useita orpo( harvinainen) sairaudet, jotka eivät pysty käsittelemään nykylääketieteen. Mahdollisuus on olemassa vain tukea näitä potilaita osittain parantamalla heidän elämänlaatuaan, mutta voittaa sairaus on mahdotonta.
Pompen tauti( yleistynyt glukogeneesiin) ei tällä hetkellä kuulu luettelo näistä sairauksista, vaikka tapoja käsitellä tätä tautia, tiede ei ole vielä löydetty. Kuitenkin Venäjän potilailla on mahdollisuus käsiteltävä omassa maassaan ja tietyin edellytyksin saada tukea valtiolta, julkiset organisaatiot ja hyväntekeväisyyssäätiöistä.
Sisältö
- Yleiset ominaisuudet
- Historia ja tilastoja
- aiheuttaa ja kehittymismekanismi
- perintökaaren
- Moderni luokitus
- oireet ja klinikan iän
- Vaiheet diagnostisia haku
- Mitä tehdä, kun diagnosoitu
- Mitä voi tarjota modernin lääketieteen
- ennusteen ja
ehkäisyYleiset ominaisuudet
Pompen oireyhtymä vaikuttaa hermoston ja lihasten solujen elin. Tämä tapahtuu puutteen vuoksi alfa-glykosideja, joka on seurausta mutaatiomuutoksia geneettisellä tasolla.
Jos potilas ei saa asianmukaista hoitoa, muutokset lihaskudoksen rakenne, jonka jälkeen se ei enää pysty toimimaan normaalisti.ajan, prosessi etenee, vaurio leviää muille lihasryhmiä, johon liittyy ankara kipu.
Vaikeissa vaiheissa potilaat menettävät kyvyn liikkua itsenäisesti ja kustannuksella lihaksen vaurioita hengityselimiä voi tulla riippuvaiseksi koneissa koneellisen ilmanvaihdon.

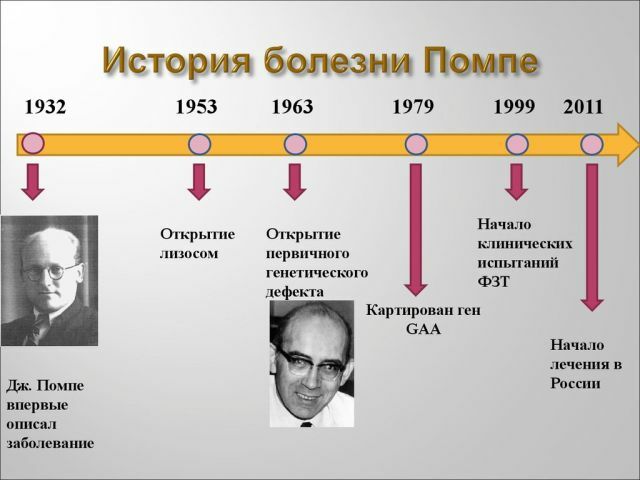
Historia ja tilastoja
paljasti tauti oli suhteellisen äskettäin - 30s viime vuosisadan. Se kuvattiin ensimmäisen kerran tutkijat Alankomaista, J. Pompen.
tauti voi lyödä samalla todennäköisyydellä molempia sukupuolia, se ei ole pääasiallisesti mies tai nainen. Varhainen muodossa tauti esiintyy 1 140000 sikiöille ja aikuisten Pompen tauti koskettaa 1 60000 ihmistä.
Vuonna 2006 menetelmien avulla tukemaan tällaisia potilaita, ja vuoteen 2013 mennessä tehokkaita tapoja löytää ja venäläiset tiedemiehet on kehitetty Yhdysvalloissa. Mutta valitettavasti varaa kohdella voi jokainen potilas hoidon hinta on hyvin korkea.
aiheuttaa ja kehittymismekanismi
taudin kehittymisen vuoksi glykogeenin talletukset luurankolihaksessa ja sydänlihaksessa. Tämä prosessi johtuu mutaatioista GAA-geenin, minkä johdosta vaikuttaa lihasten, sydänlihaksen, hermoston ja maksassa. Prosessin edetessä ja esiintyy soluissa erilaisia vaurioita, mukaan lukien dystrofinen.
Pompen tauti on yleistynyt ja lihaksikas tyyppi. Ensimmäisessä tapauksessa on muutoksia elimiin, kuten maksaan ja munuaisiin, vaikuttaa myös lihakset. Lisäksi usein ilmentymiä sydämen vajaatoiminta ja heikentynyt nielemisrefleksiä.
Kun lihas muoto oireet eivät ole niin vakavia, ja tauti ilmenee alentunut lihaskuntoa ja ryhti häiriöt. Tällaisella sairauden, potilaat turvallisesti elävät vanhoiksi.
Perintö
Pompen tautia pidetään perinnöllistä ja lähetetään autosomaalisesta - peittyvästi tyyppi. Lapsella, jonka vanhemmat ovat kantajia geenimutaatio, perinnöllinen sairaus. 
todennäköisyydellä nämä vanhemmat voivat olla lapsi Pompen oireyhtymä, on 25%.Kuitenkin mahdollisuudet ottaa suuri ja terveitä lapsia.
Science tietää jo, mitä tapahtuu ihmiskehossa, kärsivät oireyhtymä, mutta selvittää syy ei ole vielä onnistunut.
seuraa edellä esitetystä, vaarassa henkilöt, joilla on ihmisiä, joilla on samankaltainen tautien joukossa veren sukulaisia, ja mitä lähempänä sukulaisuus, sitä suurempi riskejä.Moderni luokitus
Pompen tauti perinteisesti jaettu 4 ryhmään iän mukaan, jossa ensisijainen ilmenee oireita:
- Varhainen infantile tyyppi .Taudin vakavin muoto, joka ilmenee elämän ensimmäisiltä kuukausilta vaikuttaen maksaan ja sydämeen. Yleensä lapset eivät elää vuosittain, kuolee sydämen tai hengityselinten vajaatoiminta.

- Myöhäinen lapsen tyyppi .Se ilmenee ensimmäisten kolmen vuoden elämässä ja kehittyy niin nopeasti. Potilaat kuolevat lähempänä nuoruutta, ja syy on useimmiten sydämen vajaatoiminta.
- Nuorten tyyppi .Ensimmäiset oireet ilmestyvät 6-10 vuoteen ja tappava lopputulos lähestyy 20 vuotta.
- Aikuisen sairauden tyyppi .Se ilmenee 20-40-vuotiaana, kehittyy hitaasti ja potilaalla on riittävästi mahdollisuuksia elää hyvin vanhaksi.
Taudin tutkiminen yksityiskohtaisemmin on melko vaikeaa, koska sitä pidetään suhteellisen harvinaisena.
Oireetologia ja klinikka iästä riippuen
Eri ikäryhmissä tauti ilmenee erilaisilla oireilla. Vaikein ja ohimenevä on varhaisen infantilainen sairauden tyyppi.
Taudin oireet ensimmäisinä kuukausina:
- lihasheikkous;
- matala moottoriaktiivisuus;
- viivästynyt fyysinen kehitys;
- excitability ja usein itkeminen ilman ilmeistä syytä;
- -häiriöt hengityselimissä;
- maksan laajeneminen ja sydänlihaksen leviäminen;
- nielun heikkeneminen ja imun refleksejä, kielen lisääntyminen.

Lapsia myöhäisillä lapsilla ja nuorilla( 3 - 10-vuotiaat) oireet:
- vähensi lihasääntä alaraajoissa asteittaisella siirtymällä muihin lihaksiin;
- heikentynyt liikkeiden koordinointi;
- -ongelmat hengityselimistön osalta;
- painon puute;
- lisääntyy sydän-, perna- ja maksan koossa.
Aikuisen tai myöhäisen sairauden( 20 vuoden ikä) oireet:
- unettomuus tai lisääntynyt uneliaisuus;
- usein päänsärky;
- lihasheikkous ja vaikea hengenahdistus;
- -skolioosi.
Taudin tätä muotoa pidetään vähiten vaarallisena ja kuolleisuus on paljon pienempi kuin taudin kehittyminen lapsuuteen.

Diagnostiset hakuvaiheet
 Taudin havaitseminen suoritetaan useassa vaiheessa.
Taudin havaitseminen suoritetaan useassa vaiheessa.
Ensinnäkin seuraavat tärkeimmät diagnostiset menetelmät ja sitten lisätutkimukset voidaan määrittää riippuen taudin tyypistä ja vaiheesta.
Tyypin 2 glykogenogeenin havaitsemiseen käytettävät pääasialliset menetelmät ovat:
- -anamneesi;
- -veren seerumin laboratoriotestit;
- kliinisten oireiden arviointi;
- MRI alaraajojen ja muiden kehon osien lihaksista;
- EKG- ja EchoCG-rinta röntgen.
Päädiagnostiikan suorittamisen jälkeen 
- : llä on tarkoitus tutkia DNA: ta geneettisiin vikoihin;
- -lihaskudoksen biopsia ja mikroskooppinen analyysi;
- -verikokeessa alfa-glukosidaasin aktiivisuuden tason määrittämiseksi.
Sairauden myöhäiseen muotoon sovelletaan seuraavia menetelmiä:
- polysomnografia;
- keuhkokapasiteetin määritys;
- luustolihaksen sähkömagiaografia.
Toteutettujen tutkimusten tulosten ja potilaan yleisen tilan arvioinnin perusteella määrätään hoito.
Mitä tehdä, kun diagnoosi tehdään
Joten, pettymys diagnoosi on laitettu, ja ei ole epäilystäkään. Mitä seuraavaksi? Tällöin on noudatettava seuraavia toimenpiteiden algoritmeja:
- valmistelee pakkauksen seuraavista asiakirjoista: lääkärikortin otteesta, joka sisältää tietoa diagnoosista, suositelluista lääkkeistä ja niiden käyttöönotosta;lääketieteellisten provisioiden pöytäkirjat;vammautumiskysymys;
- siirtää ilmoitettu luettelo hoitavalle lääkärille käsittelyn hakemuksen valmistelemiseksi;
- vierailee terveysviranomaisilla hakemuksella tarvittavien lääkkeiden ostamiseksi;
- kirjoittaa hakemuksen julkiselle organisaatiolle apua varten;
- , jos hakemus hylättiin, on ilmoitettava terveysministeriölle ja pyydettävä oikeusapua.
Mitä moderni lääketiede voi tarjota
 Kuten edellä mainittiin, on mahdotonta parantaa Pompe-tautia. Kuitenkin tiede on käsitellyt tätä ongelmaa useita vuosia ja tiettyjä saavutuksia on tehty. Tänään voit hidastaa prosessia ja helpottaa potilaan tilaa. Mitä modernin lääketieteen tarjonta Venäjällä tarjoaa?
Kuten edellä mainittiin, on mahdotonta parantaa Pompe-tautia. Kuitenkin tiede on käsitellyt tätä ongelmaa useita vuosia ja tiettyjä saavutuksia on tehty. Tänään voit hidastaa prosessia ja helpottaa potilaan tilaa. Mitä modernin lääketieteen tarjonta Venäjällä tarjoaa?
Ainoa venäläisten kansalaisten käytettävissä oleva lääke on Mayozaim laskimonsisäiseen antoon. Tämä on elinikäinen korjaava toimenpide, jonka tarkoituksena on palauttaa taudin kohteena olevien elinten ja järjestelmien toiminta.
Lääkettä voidaan käyttää missä tahansa ikässä ja kaikissa sairauden muodoissa. Seuraavista positiivisista vaikutuksista havaitaan sen käytön seurauksena:
- parantaa luiden ja lihasten tilaa;
- vakauttaa hengityselinten toimintaa;
- lisää moottorin toimintaa.
Mayozeim-valmisteen käyttö vähentää kuoleman riskiä 99%: lla ja parantaa hengityselinten toimintaa 92%.Joillakin potilailla voidaan kuitenkin havaita seuraavat haittavaikutukset:
- oksentelu ja pahoinvointi;
- allergiset oireet iholla;
- hengenahdistus ja takykardia.
Huumeiden haitta on sen korkea hinta. Jos hoitoa ei ole mahdollista, lääkärit korjaavat potilaan tilan halvempien ja vähemmän tehokkaiden lääkkeiden avulla.
: n ennuste ja ennaltaehkäisy Pompe-taudin ennuste riippuu taudin muodosta. Varhaisessa lapsityypissä lapset kuolevat ennen kuin he saavuttavat yhden vuoden iän. Kuoleman syy on sydämen vajaatoiminta.
Myöhäinen lapsen ja nuoren muodon ansiosta potilas voi saavuttaa 20 vuoden iän. Kuolema tapahtuu sydämen vajaatoiminnan vajaatoiminnan seurauksena.
Suotuisimpia ennusteita niille, joilla tauti diagnosoitiin 20 vuoden kuluttua. Nämä ihmiset voivat elää vanhuksille, mutta lähes jokaisella on vamma.
Ennaltaehkäisyä ei ole olemassa. Tämä on geneettinen sairaus, eikä ole olemassa takeita siitä, että terve lapsi syntyy perheessä, jossa on ollut samanlaisia sairauksia. Voit määrittää vain todennäköisen sairastuvuustodennäköisyyden asteeseen suorittamalla asianmukainen tutkimus.