les mariages associés ne font qu'accroître le nombre d'enfants nés avec ce diagnostic. Le plus fréquemment observé dans le nord de l'Europe phénylcétonurie - 1: 10.000, en Russie 1: 8-10000 et en Irlande - 1: 4560. En phénylcétonurie noir presque ne se produit pas.

Quelle est cette maladie?
Phénylcétonurie - un fermentopathia groupe de maladies héréditaires associées à une altération du métabolisme des acides aminés, principalement phénylalanine. Le non-respect régime pauvre en protéines est accompagnée par l'accumulation de phénylalanine et de ses produits toxiques, ce qui conduit à lourde défaite du système nerveux central qui se manifeste notamment sous la forme de violations du développement intellectuel (phénylpyruvique un retard mental). L'un des rares maladies héréditaires qui se prêtent à un traitement efficace.
histoire
Phénylcétonurie a ouvert en 1934 un médecin norvégien Ivar Asbjørn Fölling. Le résultat positif du traitement a été observé dans le (Birmingham Hospital for Children) au Royaume-Uni grâce aux efforts déployés par l'équipe de médecins dirigée par Horst Bickel dans la première moitié des 50-s du XX siècle. Mais le succès vraiment grand dans le traitement de la maladie a été enregistrée dans les années 1958-1961, lorsque le premier les méthodes d'analyse du sang de nourrissons à son contenu de concentrations élevées de phénylalanine, en parlant de la présence maladie.
Il est avéré que pour le développement de la maladie est responsable un seul gène, a reçu le nom de la RAS (phénylalanine gène hydroxylase).
En raison de cette découverte, les scientifiques et les médecins à identifier pourraient mieux dans le monde entier et décrire à la fois la maladie elle-même et de ses symptômes et les formes. En outre, il a été trouvé et mis au point une nouvelle méthodes complètement, high-tech et modernes de traitement, tels que la thérapie génique, qui est aujourd'hui un exemple d'un contrôle efficace des pathologies génétiques personne.

Le mécanisme de développement et les causes de la maladie
La cause de cette maladie est associée avec le fait que l'enzyme ne se produit pas dans le foie humain - phénylalanine 4-hydroxylase. Il est responsable de la conversion de la phénylalanine en tyrosine. La dernière partie de la mélanine, pigment, les enzymes et les hormones nécessaires pour la fonction normale du corps.
Lorsque phénylcétonurie phénylalanine, ce qui, en échange de façons défavorable, transformé en substance, ce qui devrait être dans le corps: et phényl- acide phénylpyruvique et ortofenilatsetat phényléthylamine. Ces composés s'accumuler dans le sang et ont une action complexe:
- violer les processus du métabolisme des graisses dans le cerveau;
- avoir un effet toxique, l'empoisonnement du cerveau;
- causer une carence des neurotransmetteurs qui transmettent l'influx nerveux entre les cellules du système nerveux.
Cela provoque une réduction significative et irréversible dans l'intelligence. Le bébé se développe rapidement un retard mental - un retard mental.
La maladie est héréditaire que lorsque les deux parents donnent à l'enfant une tendance à la maladie, et donc tout à fait rare. Deux pour cent des personnes ont le gène modifié responsable de la maladie. Cet homme est tout à fait sain. Mais quand un homme et une femme, les porteurs du gène muté, se marier et décider d'avoir des enfants, la probabilité que les enfants souffriront de phénylcétonurie, est de 25%. Et la possibilité que les enfants seront porteurs du gène anormal de phénylcétonurie, mais ils restent relativement en bonne santé, est de 50%.
symptômes de phénylcétonurie
Phénylcétonurie (voir. photo) apparaît dans la première année de vie. Les principaux symptômes de cet âge sont les suivants:
- bébé léthargie;
- vomissements;
- troubles du tonus musculaire (hypotonie souvent);
- convulsions;
- le manque d'intérêt dans les environs;
- parfois l'irritabilité;
- l'anxiété;
- signes de dermatite allergique;
- il y a une odeur « de la souris » caractéristique de l'urine.
Si phénylcétonurie est caractérisé par les caractéristiques phénotypiques suivantes: dépigmentation de la peau, des cheveux et de l'iris. Chez certains patients, l'une des manifestations de la maladie peuvent être sclérodermie.
Dans la vie plus tard pour les patients atteints de phénylcétonurie sont caractérisés par un retard de développement mental et de la parole, souvent marqué microcéphalie. Les crises d'épilepsie se produisent dans près de la moitié des patients atteints de phénylcétonurie et dans certains cas, peut être le premier signe de la maladie.
diagnostic
Important, comme nous l'avons déjà noté, est le diagnostic précoce de la maladie, afin d'éviter son développement et conduire à un certain nombre d'effets irréversibles et graves. Pour cette raison, dans les services de maternité à 4-5 jours de la vie (pour enfants nés à terme) pris pour l'analyse du sang. Les bébés prématurés de la phénylcétonurie (PKU) sang pris à 7 jours.
La procédure consiste à prélever du sang capillaire après heure après le repas, en particulier, elle est imprégnée d'une forme particulière. La concentration, ce qui indique la marque de plus de 2,2% de phénylalanine dans le sang du bébé a besoin de directives de ses parents pour l'inspection dans le Centre de génétique médicale. Il y a aussi procédé à un examen plus approfondi et, en fait, un diagnostic précis.
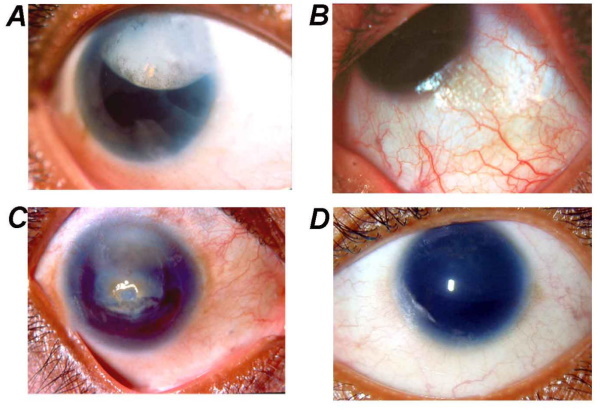
Qu'est-ce que phénylcétonurie: photo
La photo ci-dessous montre comment la maladie se manifeste chez les enfants et les adultes.

Comment traiter phénylcétonurie
Le seul traitement efficace de la phénylcétonurie est considérée comme organisée depuis les premiers jours de régime de vie spécialement formulé, dont le principe est de limiter la phénylalanine contenue dans les aliments, qui a éliminé ces aliments comme:
- céréales,
- légumineuses,
- oeufs,
- fromage cottage,
- produits de boulangerie,
- noix,
- chocolat,
- le poisson, la viande et ainsi de suite.
alimentation thérapeutique des patients atteints de phénylcétonurie se compose de produits spécialisés, la production étrangère et domestique. Les enfants la première année de la vie montrent les produits dans sa composition proche du lait maternel, il est un tel mélange comme « Lofenilak » et « Afenilak ». Pour les enfants un peu plus développé que ces mélanges « Tetrafen », « Maksamum-XP », « Phenyl-libre. » PKU les femmes enceintes et les enfants plus âgés (après six ans) montre un mélange typique « Maksamum-XP ». En plus des produits médicaux spécialisés dans l'alimentation du patient des jus, des fruits et légumes.
diétothérapie en temps opportun lancé évite souvent le développement des manifestations cliniques caractéristiques de phénylcétonurie classiques. Le traitement est effectué sans échec avant la puberté, et parfois plus. Parce que les patients avec la femme de phénylcétonurie faire un fœtus sain n'est pas capable, maintenu avant même la conception commencé et en continuant jusqu'à le traitement spécial des naissances à l'exclusion de la phénylalanine fœtale du patient mère.
Sous les enfants Le traitement doit être sous la surveillance constante d'un pédiatre et neuropsychiatre. Au début du traitement du contrôle du contenu de phénylcétonurie phénylalanine est effectuée sur une base hebdomadaire, à la normalisation taux de conversion à 1 fois par mois pendant la première année de la vie, et 1 fois en deux mois chez les enfants plus âgés année.
En plus du traitement de l'alimentation, les enfants avec les médecins de phénylcétonurie peuvent faire les nominations suivantes:
- des composés minéraux;
- nootropics;
- vitamines B;
- anticonvulsivants.
Dans le traitement doit assister à la physiothérapie, l'acupuncture et le massage.
Remarque: quand une forme atypique de phénylcétonurie, qui ne peut être corrigée diétothérapie, les médecins prescrivent gepatoprotektory, anticonvulsivants. Ce traitement aidera à soulager la condition de l'enfant.
Phénylcétonurie et de la maternité
Pour les femmes enceintes, les patients atteints de phénylcétonurie, il est très important de garder un faible niveau de phénylalanine avant et pendant la grossesse, afin que l'enfant était en bonne santé. Bien que, seul le développement du fœtus peut être porteur du gène phénylcétonurie mais l'environnement intra-utérin peut avoir un niveau très élevé de phénylalanine, qui a la capacité de traverser le placenta. En conséquence, le bébé peut développer une maladie cardiaque congénitale, un retard de développement possible, microcéphalie et un retard mental. En règle générale, les patients avec les femmes phénylcétonurie aucune complication pendant la grossesse ne se produit pas.
Dans la plupart des pays, les femmes atteintes de phénylcétonurie qui envisagent d'avoir des enfants, il est recommandé de réduire le niveau de phénylalanine (Habituellement jusqu'à 2-6 mol / l) avant la grossesse, et pour le contrôler au cours de la période de gestation enfant. Ceci est réalisé par des tests sanguins réguliers et l'adhésion à un régime strict et une surveillance constante d'une diététicienne. Dans de nombreux cas, dès que le foie du foetus commence à produire des HAP normalement, le niveau de phénylalanine le sang de la mère tombe, respectivement, « il est nécessaire » pour l'augmenter, de maintenir un niveau de sécurité - 2-6 mol / l.
Voilà pourquoi, la quantité consommée quotidiennement par la mère de phénylalanine peut doubler, voire tripler d'ici la fin de la grossesse. Si le niveau de phénylalanine dans le sang de la mère est inférieure à 2 mol / litre, parfois, les femmes peuvent éprouver divers complications associées à une carence de cet acide aminé, tels que des maux de tête, nausées, perte de cheveux et un général malaise. Si de faibles niveaux de phénylalanine chez les patients atteints de PCU est maintenue tout au long de la grossesse, le risque d'avoir un enfant malade n'est pas plus élevé que celui de ces femmes qui ne sont pas malades phénylcétonurie.
prévention
Depuis phénylcétonurie - une maladie génétique, il est impossible d'empêcher complètement le développement. Les mesures de prévention visant à prévenir les troubles graves irréversibles du développement du cerveau grâce à un diagnostic en temps opportun et la thérapie alimentation.
Les familles qui ont déjà eu des cas de cette maladie, il est recommandé d'effectuer une analyse génétique qui peut prédire le développement possible de l'enfant a phénylcétonurie.
Conséquences et perspectives de la vie
L'exposition à des quantités excessives de phénylalanine sur le système nerveux de l'enfant a conduit à des troubles psychologiques persistants. À l'âge de 4, sans traitement approprié, les patients ayant des enfants de phénylcétonurie sont classés en tant que membres débiles et physiquement sous-développés de la société. Ils rejoignent les rangs de l'enfance et les couleurs de la vie des personnes handicapées pour les estomper.
Il ne brille pas avec le bonheur et la vie des parents d'un enfant malade. Enfant nécessite des soins constants, et avec des ressources financières limitées, cela se traduit par une détérioration totale de la richesse familiale. La douleur ressentie par la mère et le père de l'incapacité à changer la vie de l'enfant pour les meilleurs opprime et broie, mais ne peut pas être découragé. Aidez-vous, aider votre enfant à passer par ces épreuves avec moins de perte d'amour et de miséricorde.
La science à la hâte, il fait les progrès de sept lieues vers l'élimination de la maladie de qualité lourde. Il est crucial de diagnostic phénylcétonurie dans l'utérus, mais si une telle méthode n'a pas été inventé. « Pourtant, » ne veut pas dire « jamais », nous allons attendre et croire