 Voor de eerste keer werd het Louis-Bar-syndroom in 1941 in Frankrijk gezien en beschreven. Sindsdien is zijn verschijningsfrequentie merkbaar toegenomen en begon hij overal ter wereld te verschijnen.
Voor de eerste keer werd het Louis-Bar-syndroom in 1941 in Frankrijk gezien en beschreven. Sindsdien is zijn verschijningsfrequentie merkbaar toegenomen en begon hij overal ter wereld te verschijnen.
Statistieken zeggen dat in de huidige maatschappij 1 op de 40 duizend mensen de kans heeft om dit syndroom te hebben.
De essentie ligt in de aangeboren abnormale immuuntoestand van het lichaam , die met name de T-link beïnvloedt en zich begint te manifesteren in abnormale veranderingen door het hele lichaam.
Mensen met het syndroom zijn vatbaar voor frequent voorkomende infectieziekten en hebben ook een grote kans kwaadaardige tumoren door het hele lichaam te ontwikkelen.
Meestal, als het Louis-Bar-syndroom al bij de geboorte bij kinderen begint te verschijnen, dan zit het vol met dodelijke slachtoffers, zelfs zonder de kans om zo'n patiënt correct en op tijd te diagnosticeren.
Ziekte in dezelfde verhouding treft zowel mannen als vrouwen zo snel mogelijk en vernietigt hun zenuwstelsel en huid.
Oorzaken van
Het syndroom kan plaatsvinden op genetisch niveau, met de geringste storing of afwijking.
Een soortgelijke fout zit vol met neuro-ectodermale dysplasie, die bij deze mensen ingeboren is.
Pathologie verwijst naar autosomale recessieve ziekten, die kunnen optreden als de genaandoeningen tegelijkertijd bij beide ouders aanwezig waren.
De ziekte heeft de neiging om de weefsels van het cerebellum volledig te veranderen en te vernietigen, zelfs tot in de kern.
Vergelijkbare situaties leiden tot degeneratieve veranderingen in de hersenschors, evenals het ruggenmerg.
Het is mogelijk om het alleen te laten zien na een lange en moeilijke behandeling van infectieziekten, die niet het gewenste resultaat geven.
Sterke immuunstoornissen leiden tot de vorming van kwaadaardige tumoren, die hun oorsprong vinden in het lymforeticulaire systeem.
Symptomen van
-syndroom In de moderne geneeskunde is pathologie zeldzaam, maar artsen zijn bang voor de mogelijke ontwikkeling van 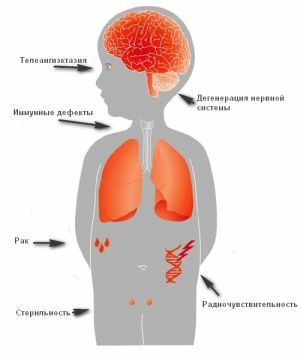 .
.
Aangezien deze genetische ziekte cellulaire immuniteit gedeeltelijk of volledig vernietigt, heeft het een pathologisch karakter en kan het niet worden behandeld. Een volledig leven is bijna onwerkelijk.
Symptomatische ziekte op volwassen leeftijd kan niet onmiddellijk worden uitgedrukt.
Meestal wordt het gedetecteerd door de geleidelijke verslechtering van de interne organen, de nederlaag van het immuunsysteem, volledige of gedeeltelijke afwezigheid van de thymus.
Als Louis-Bar syndroom ontwikkelt zich in de baarmoeder, waardoor de kleine hersenen en cerebrale cortex kind, pasgeboren vanaf de geboorte van een degeneratieve verandering en veroordeelt de diagnose kwellen.
Als de baby bij de geboorte de eerste tekenen van de ziekte niet heeft opgemerkt, zal het syndroom op de leeftijd van 3-24 maanden vrij snel gaan manifesteren.
de meeste gevallen leidt dit tot de volledige afwezigheid van bewegingen, slechte coördinatie, en cognitieve ontwikkeling stagnatie uiterlijke kenmerken van het gezicht en ledematen.
Het kan zijn:
- -spier hypotensie;
- scheelzien;
- gebrek aan reflexen en functionaliteit van spieren en ogen.
Het Louis-Bar-syndroom manifesteert zich vaak in aanhoudende infectieziekten die de luchtwegen en oren betreffen.
Dit kan otitis media zijn, faryngitis, bronchitis, sinusitis en andere ziekten.
Longontsteking en pneumonie komen bijna nooit tot uiting. Elke volgende ziekte heeft een meer acute vorm en complicaties die niet vatbaar zijn voor behandeling.
Het teken van het symptoom wordt ook beschouwd als vasculair sterretje, dat op het derde levensjaar door het lichaam kan verschijnen.
Meestal is dit te wijten aan de uitbreiding van de haarvaten, maar als er slechts een symptoom van de noodzaak om te kijken naar andere opties van mogelijke ziekten.
Wat betreft het uiterlijk van het gezicht en de ogen, hier in de eerste plaats begint te telangiectasia manifesteren op de oogbol.
is beladen met een constante conjunctivitis, visuele symptomen die zich kunnen manifesteren niet alleen voor, maar ook op de hals, wangen, oren, oogleden en zelfs op de handpalmen.
Naast deze code wordt het hele lichaam droog en schilferig, de haarbedekking is overvloedig.
In de meest verwaarloosde situaties kan het syndroom kwaadaardige tumoren, leukemie en lymfoom veroorzaken.
Wat is er gedaan voor de diagnose?
 Bij het eerste teken of vermoeden van een dergelijke ziekte, maakt elke arts een afspraak en verwijst hij door naar een arts met een nauwere specialisatie.
Bij het eerste teken of vermoeden van een dergelijke ziekte, maakt elke arts een afspraak en verwijst hij door naar een arts met een nauwere specialisatie.
Heel vaak worden dergelijke patiënten gelijktijdig waargenomen door verschillende artsen die de behandeling voorschrijven in een gezamenlijk consult.
Dit kan een immunoloog, dermatoloog, oogarts, neuroloog, oncoloog en een audioloog zijn. Alleen hun gezamenlijk overleg zal dit symptoom kunnen onderscheiden van andere zeldzame en gevaarlijke soorten ziekten.
De uiteindelijke diagnose bij een dergelijke ziekte wordt altijd alleen door een neuroloog gesteld als hij alle resultaten van klinische tests en laboratoriumtests bij de hand heeft.
Meestal helpen sommige indicatoren om de diagnose vast te stellen, die niet overeenkomt met de norm. In het bijzonder kan het bloed volledig lymfocyten missen, en het niveau van immunoglobuline zal veel lager zijn dan normaal.
Tegelijkertijd zullen absoluut geen antilichamen beschikbaar zijn om virale infecties en ziekten te bestrijden.
Wanneer de definitieve diagnose voor een neuroloog op zijn handen is, dan kunt u een specifiek verloop en behandelingsregime voor een dergelijke patiënt voorschrijven.
Hoe kan het leven van een patiënt worden verlengd?
Helaas heeft het niveau van de geneeskunde helaas niet het niveau bereikt om effectieve en snelle methoden te vinden om deze genetische aandoening te bestrijden.
De behandelingsmethoden zijn nog steeds onderwerp van het onderzoek en de studie van veel wetenschappers. Om de levensondersteuning van dergelijke patiënten te behouden, is het echter gebruikelijk om palliatieve symptomatische behandeling te gebruiken. 
Om de levensduur van deze patiënten langer specifieke immuuntherapie, die verschillende doseringen van T-activine en gammaglobuline kunnen voorschrijven.
Tegelijkertijd is een constante hoge dosis vitaminepreparaten verplicht, die op een complexe manier worden geïntroduceerd om de goede werking van het hele organisme te behouden.
Als de patiënt met het syndroom van Louis Bara heeft een aantal besmettelijke ziekte, wordt deze behandeld op de intensive care in de eerste plaats om het proces van het handhaven van het lichaam op het juiste niveau zonder al te veel bacteriën en virussen te starten.
Afhankelijk van de stoornissen die in het lichaam worden waargenomen, kunnen geneesmiddelen en hun doseringen aanzienlijk variëren. Vaak wordt de loop van de therapie aangevuld met antischimmel- en antivirale middelen, evenals sterke antibiotica.
 Ongeoorloofd en onredelijk opblazen van de psyche over niet-bestaande ziekten - hypochondrische neurose. Wat moet er gedaan worden om de stoornis te behandelen?
Ongeoorloofd en onredelijk opblazen van de psyche over niet-bestaande ziekten - hypochondrische neurose. Wat moet er gedaan worden om de stoornis te behandelen? Dan Jackson's epilepsie verschilt van andere soorten ziekten. Methoden voor diagnose en behandeling worden hier besproken.
realistische prognoses
Zoals Louis-Bar syndroom is vrij nieuw en totaal onontgonnen, dan praten over een grote kans van de behandeling en nog meer op het herstel van de patiënt is onmogelijk.
De pathologie heeft een ongunstige prognose, die, afhankelijk van verschillende factoren, beide vele jaren op hetzelfde niveau kan plaatsvinden en snel kan worden afgebouwd.
Het meest voorkomende symptoom is te vinden in een diepe kindertijd of bij de geboorte van een kind. De gemiddelde leeftijd van deze kinderen is ongeveer 3 jaar.
Als de symptomen later aan het licht komen, overleven dergelijke patiënten tot een maximum van 20 jaar.
Meestal is de oorzaak van hun dood niet de ziekte zelf, maar de volledige vernietiging van immuniteit en de snelle ontwikkeling van oncologische formaties door het hele lichaam.