 degenerative-dystrofiske sykdommer i nervesystemet med en overveiende involvering av de perifere nerver og muskelfibrene oppta en stor andel i strukturen av humane arvelige sykdommer.
degenerative-dystrofiske sykdommer i nervesystemet med en overveiende involvering av de perifere nerver og muskelfibrene oppta en stor andel i strukturen av humane arvelige sykdommer.
typisk representant er myotonic dystrofi( eller dystrofe myotoni) beskrevet i begynnelsen av forrige århundre, flere forfattere og kalt sykdom Rossolimo-Steinert-Kurshmana.
Denne sykdommen er den mest kjente av sykdommen myotonic utslipp og den vanligste formen for muskeldystrofi hos voksne. Hva er denne sykdommen og hvordan bekjempe den?
Åpning og er
Rossolimo sykdom, Steinert og Kurshmana studert sykdom er genetiske lidelser med autosomal dominant arv. Dette betyr at en forelder har et mutantgen, syke barn er født med en sannsynlighet på 50%.Sykdommen har karakter av en familieproblemer og overføres til etterfølgende generasjoner i vertikal skala.
sønner og døtre i disse familiene lider med samme frekvens, ca 3-5 personer per 100 000 innbyggere. Alderen på sykdomsutbruddet, så vel som alvorlighetsgraden av symptomene, er merkbart variabel.
beskrive tidlige neonatale og sene former, men ofte sykdommen gjør sin debut på den andre, minst - i tredje tiår av livet.  bemerket at overføring av sykdommen fra mor til barn er mer prognostisk ugunstig enn fra sin far.
bemerket at overføring av sykdommen fra mor til barn er mer prognostisk ugunstig enn fra sin far.
Grunnlaget for gen feil sykdom er over 19 par kromosomer som er ansvarlig for syntesen av enzymet proteinkinase-miotonin. Dette proteinet er normalt til stede ikke bare i skjelettmuskel, men også i cellene i myokard og sentralnervesystemet.
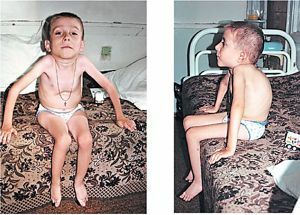
Det er derfor dystrophic myotoni karakteristiske polisistemny manifestasjoner med nederlaget av ulike organer og systemer. Mindreverdighets miotonin-kinase fører til muskelspasmer, sammen med atrofiske forandringer i muskulaturen av hodet, nakkesøylen, ekstremiteter. Det
kombinasjon hypertrofi av muskelfibre med noen annen atrofi og erstatte dem på fett eller bindevev.
Kliniske manifestasjoner
Grunnet variasjonen av utbruddet av sykdommen i klinikken er følgende former etter alder:
- medfødt skjema - en manifestasjon av sykdommen begynner umiddelbart etter fødselen av barnet,
- ungdommelig versjon - myotoni debut i en alder av ett år til pubertet;
- klassiske form - utbruddet av kliniske manifestasjoner har andre og tredje tiår av livet;
- minimal versjon - demonstrasjonskonto for senere perioder - sjette tiår av livet.
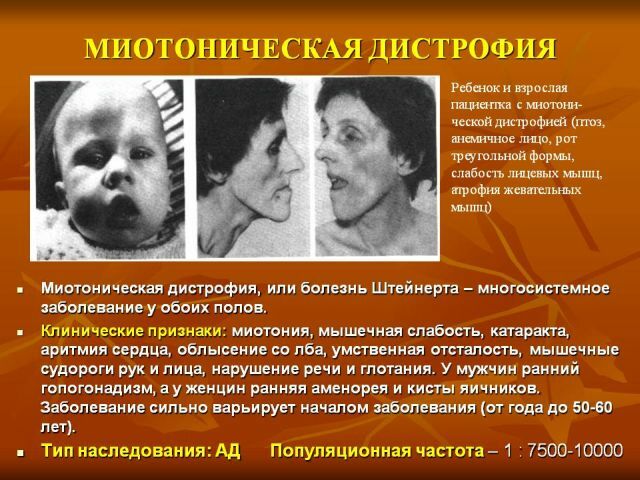
Karakteristisk, jo senere manifesterer sykdommen, jo mer gunstig kurset og jo bedre prognosen. Den mest vanlige klassiske form av Steinert sykdom, for hvilke de følgende kliniske tegn er typiske:
- myotoni - manifestert ved kramper i tygge muskler og bøyemuskler i hendene, kjennetegnet ved atrofiske endringer i ulike
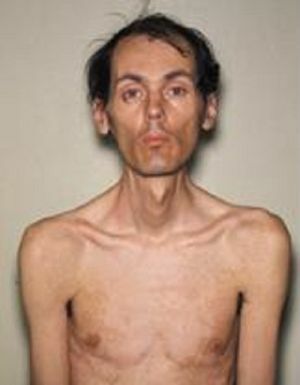 muskelgrupper. Gradvis fading myotonisk symptomer og progresjon av muskulær dystrofi, utvendig det uttrykkes i en trist ansiktsmasken og ansikts fravær. Farlig er parese av musklene i strupehode med svekket svelge, og svakhet i åndedrettsmuskulaturen, noe som resulterer i mulige angrep av apnea under søvn, utvikling av lungebetennelse.
muskelgrupper. Gradvis fading myotonisk symptomer og progresjon av muskulær dystrofi, utvendig det uttrykkes i en trist ansiktsmasken og ansikts fravær. Farlig er parese av musklene i strupehode med svekket svelge, og svakhet i åndedrettsmuskulaturen, noe som resulterer i mulige angrep av apnea under søvn, utvikling av lungebetennelse. - Kardiovaskulære sykdommer - hjertearytmier, hypertrofiske endringer av venstre ventrikkel, påvist på et elektrokardiogram, kongestiv hjertesvikt.
- endokrine lidelser( hovedsakelig påvirket seksuell funksjons) - minskning i størrelsen på kjønnsorganer, nedsatt seksuell lyst hos kvinner - menstruasjonsforstyrrelser, fedme.
- generelle endringer dystrofe - tørrhet og pigmentering av huden, tap av deler av eller hele håret og tennene, tidlig grå stær.
- CNS bivirkninger - tretthet, søvnforstyrrelser, apati, tap av intelligens.
Separat verdt å merke seg typiske kliniske manifestasjoner av medfødt former dystrofe myopati:
- reduksjon i aktive føtale bevegelser i livmor, oppdaget under ultralyd;
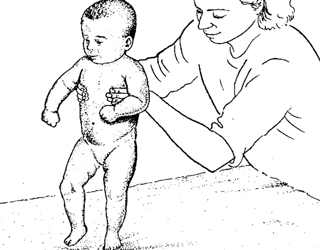
- i nyfødtperioden - apati, hypotensjon utbredt, spesielt i tygge, ansiktsbehandling, musklene i øyeeplet;
- bevaring og jevn økning av tendonreflekser;
- fôrproblemer, åndedrettsstress som åndedrettssyndrom;
- forsinkelse av fysisk og nevropsykisk utvikling, tegn på oligofreni;
- rask sykdomsprogresjon, høy risiko for plutselig død.
Diagnostiske kriterier
mistanke om sykdom Rossolimo-Steinert - Kurshmana kan forekomme i legen om pasienten har en kombinasjon av myotonisk dystrofi og endringer i musklene på bakgrunn av tap av intelligens og nærvær av kardiovaskulære og endokrine sykdommer.
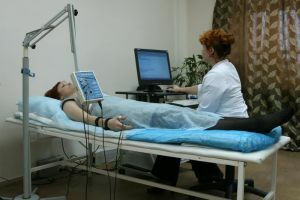
Polysystemisk indikerer nesten alltid genetisk natur av sykdommen. Slike pasienter utsatt for DNA-analyse og føring genealogical analyse for å bekrefte arv av en autosomal dominant sykdom. Som informative metoder for forskning, elektrokardiografi, elektroneuromyografi, hormontester, brukes.
På grunn av allsidigheten av de kliniske manifestasjoner av prosessen med diagnosen er vanligvis involvert spesialister fra forskjellige grener av medisin - genetikk, kardiologi, endokrinologi, gynekologi, Andrologi Neurology.
differensialdiagnose mellom dystrofisk myotoni og andre typer lignende sykdommer. Til forskjell fra resten er muskelatrofi karakteristisk for Rossolimos sykdom. Ofte, for å bekrefte diagnosen er nødvendig å ty til en biopsi for å bestemme nivået av muskelprotein, som i vevene i denne patologi økt.
Antenatal diagnose utføres også ved hjelp av metoden for fostervannforskning.
medisinsk assistanse
genetisk sykdom kan ikke helbredes fullstendig, slik at målet for behandlingen i sykdoms Rossolimo-Steinert-Kurshmana er symptomlindring, forbedring i den generelle tilstand og sosial tilpasning av pasientene.
behandlingsprinsipper er som følger:
- diett lav kaliumsalter( epler, asparges, kål, agurk, drue, grønnsaker, mais, bær, reddiker, mandariner, grapefrukt
 , løk, gulrøtter, aubergine, ert);
, løk, gulrøtter, aubergine, ert); - undertrykkelse av superkjøling for å unngå spasmer;
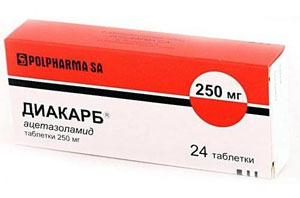
- bruk av narkotika kinin for å stabilisere cellemembraner på slike medikamenter som Difenin, prokainamid, Diakarb - for å lindre muskelspasmer og redusert stivhet, beslag, reduserer intrakranialt trykk;
- bruk av anabole steroider ( Metanandrostenolon, Retabolil, Nerabol), vitaminer, ATP for å stimulere muskelmasse;
- treningsterapi, massasje, elektro, ortopediske enheter.
Disse arrangementer gir en god positiv effekt både på klassisk og i medfødt form av sykdommen. Fullt avlaste pasienten som de ikke kan dystrofe myotoni, men å forlenge livet og forbedre kvaliteten kan.
dårligere prognose i medfødte former - dødeligheten er høy, barn kan ikke leve opp til 3 år. Junior versjon av myotoni oppstår 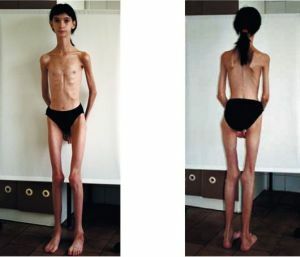 ganske vanskelig og kan føre allerede i ung alder for å begrense uførhet og tidlig uførhet.
ganske vanskelig og kan føre allerede i ung alder for å begrense uførhet og tidlig uførhet.
I tilfelle av den klassiske formen av sykdommen kan oppstå lang tid på å gjennomføre rettidig behandling og rehabiliterende aktiviteter. Den mest gunstige prognosen i sykdomsproblemer med sen oppstart.
Forebyggende tiltak er redusert til det faktum at kvinner fra vanskeligstilte familier med en historie i planleggingsfasen av svangerskapet er nødvendig for å bli undersøkt for tilstedeværelse av unormale gener ansvarlig for utviklingen av muskeldystrofi. Det er også tilrådelig å gjøre dette dersom patologen til fars slektninger er tilstede.
muligheter for fødselen av barn må behandles individuelt i hvert enkelt tilfelle, legen - genetikk etter konsultasjon.