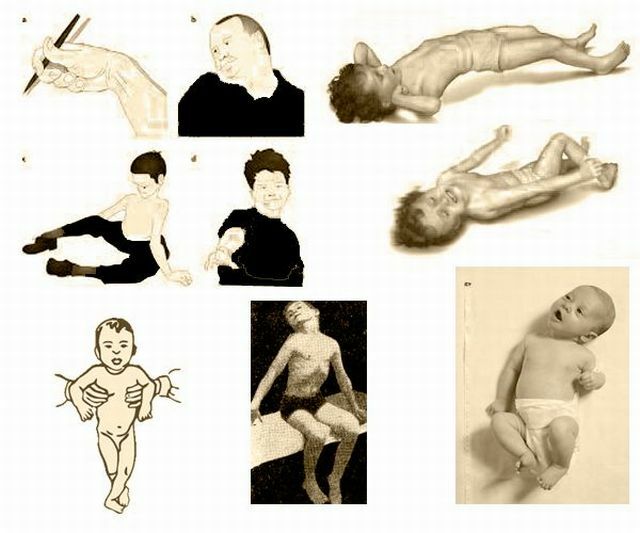
Obecnie istnieje szereg sierocych( rzadko) chorób, które nie są w stanie poradzić sobie współczesnej medycyny. Istnieje możliwość wspierania takich pacjentów, w pewnym stopniu poprawiając ich jakość życia, ale choroba nie udaje się.
Obecnie istnieje szereg sierocych( rzadko) chorób, które nie są w stanie poradzić sobie współczesnej medycyny. Istnieje możliwość wspierania takich pacjentów, w pewnym stopniu poprawiając ich jakość życia, ale choroba nie udaje się.
Choroba Pompego( uogólnione glikogenezą) aktualnie nie znajdują się na liście tych chorób, chociaż sposoby leczenia tej choroby, nauka jeszcze nie znaleziono. Jednak pacjenci rosyjskie mają możliwość być traktowane we własnym kraju i pod pewnymi warunkami, otrzymać pomoc od państwa, organizacji publicznych i fundacje charytatywne.
- Content Ogólna charakterystyka
- Historia i statystyka
- przyczyny i mechanizm rozwoju
- Dziedziczenie
- objawów Nowoczesna klasyfikacja
- i kliniki w zależności od
- wiekowej Stages diagnostyczny wyszukiwarki
- Co zrobić, gdy zdiagnozowano
- Co może zaoferować nowoczesne prognozę medycyna
- i zapobiegania
Ogólna charakterystyka zespołu
Pompego wpływa na nerwy i komórki mięśniowe ciała. Dzieje się tak ze względu na brak alfa-glikozydy, które jest wynikiem mutacji zmiany na poziomie genetycznym.
Jeżeli pacjent nie otrzymuje właściwego leczenia, zmiany struktury tkanki mięśniowej, po którym nie jest już w stanie normalnie funkcjonować.Proces postępuje z czasem, zmiana rozprzestrzenia się na inne grupy mięśni, czemu towarzyszy silny ból.
W ostrych fazach pacjenci tracą zdolność do przemieszczania się niezależnie i kosztem uszkodzenia mięśnia w układzie oddechowym mogą się zależna od maszyny do wentylacji mechanicznej.

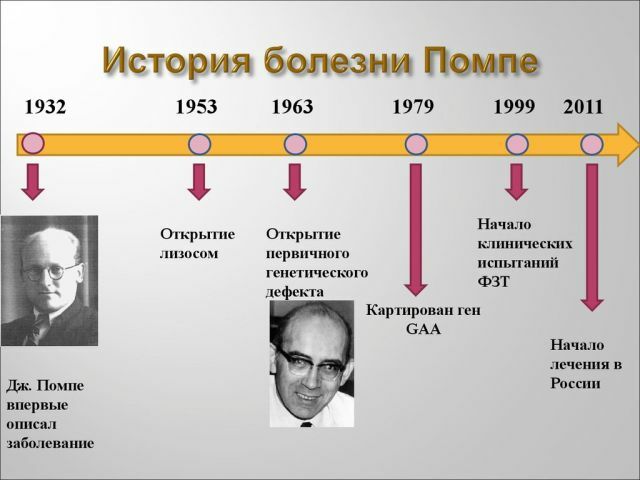
Historia i statystyka
ujawnił choroba była stosunkowo niedawno - w latach 30-tych ubiegłego wieku. Po raz pierwszy został opisany przez naukowca z Holandii, I. Pompe. Choroba
stanie trafić z równym prawdopodobieństwem obu płci, nie jest przeważnie płci męskiej lub żeńskiej. Wczesna postać choroby występuje u 1 na 140.000 do nienarodzonych dzieci i dorosłych choroba Pompego dotyka 1 na 60000 osób.
W 2006 roku metodologia pozwala na wsparcie takich pacjentów, a do roku 2013 skutecznych sposobów, aby znaleźć i rosyjscy naukowcy została opracowana w Stanach Zjednoczonych. Niestety nie każdy pacjent może sobie pozwolić na leczenie, ponieważ koszt terapii jest bardzo wysoki.
przyczyny i mechanizm rozwoju choroby
rozwija się z powodu osadów glikogenu w mięśniach szkieletowych i mięśniu sercowym. Proces ten jest wynikiem mutacji w genie GAA, w wyniku czego mięśnie Podlegające, mięsień serca, układ nerwowy i wątrobę.Proces postępuje, aw komórkach występują różne rodzaje uszkodzeń, w tym dystrofia.
Choroba Pompego jest uogólniona i muskularna. W pierwszym przypadku są zmiany w narządach, takich jak wątroba i nerki, również dotknięte układu mięśniowego. Ponadto, objawy niewydolności serca i naruszenie odruchu połykania nie są rzadkie.
Kiedy objawy kształt mięśni nie są tak poważne, a choroby objawia się obniżone napięcie mięśni i pozycja zaburzeń.Przy tej chorobie pacjenci spokojnie żyją do późnej starości. Choroba
Dziedziczenie
Pompego jest uważana za dziedziczną i jest przesyłany jako autosomalny recesywny - typu. Dziecko, którego rodzice są nosicielami genu mutacyjnego, dziedziczy chorobę.
prawdopodobieństwo, że ci rodzice mogą mieć dziecko z zespołem Pompego, wynosi 25%.Istnieją jednak duże szanse na urodzenie zdrowych dzieci.
Nauka już wie, co dzieje się w organizmie człowieka, cierpiącego na zespół, ale znaleźć przyczynę jeszcze nie udało.
Wynika z powyższego, w grupie ryzyka, którzy mają osoby z podobnym choroby wśród krewnych, a im bliżej pokrewieństwa, tym większe ryzyko. Nowoczesne klasyfikację choroby
Pompego konwencjonalnie podzielone na 4 grupy w zależności od wieku, w którym podstawowy przejawia objawów:
- Wczesny infantylny typ .Najcięższa postać choroby, która objawia się od pierwszych miesięcy życia, wpływając na wątrobę i serce. Z reguły dzieci nie żyją do roku, umierając z powodu niewydolności serca lub układu oddechowego.

- Późny typ infantylny .Przejawia się w pierwszych 3 latach życia i rozwija się nie tak szybko. Pacjenci umierają bliżej dorastania, a przyczyną jest najczęściej niewydolność serca.
- Typ młodzieńczy .Pierwsze objawy pojawiają się od 6 do 10 lat, a śmiertelny wynik zbliża się do 20 lat.
- Typ choroby u osoby dorosłej .Przejawia się ona od 20 do 40 lat, rozwija się powoli, a pacjent ma wystarczające szanse, aby żyć do bardzo starości.
Bardziej szczegółowe badanie choroby jest dość trudne, ponieważ uważa się ją za stosunkowo rzadką.
Symptomatologia i klinika w zależności od wieku
W różnym wieku choroba objawia się różnymi objawami. Najcięższy i najbardziej ulotny jest wczesny rodzaj infekcji.
Objawy choroby w pierwszych miesiącach życia:
- osłabienie mięśni;
- niska aktywność motoryczna;
- opóźniony rozwój fizyczny;
- pobudliwość i częsty płacz bez wyraźnego powodu;
- zaburzenia w układzie oddechowym;
- powiększenie wątroby i proliferacja mięśnia sercowego;
- naruszenie połykania i odruchów ssania, wzrost języka.

Objawy późnej postaci dziecięcej i młodzieńczej( od 3 do 10 lat):
- zmniejszył napięcie mięśniowe kończyn dolnych stopniowo przechodząc na inne mięśnie;
- zaburzenie koordynacji ruchu;
- problemy ze strony układu oddechowego;
- brak masy;
- zwiększenie wielkości serca, śledziony i wątroby.
Objawy postaci dorosłej lub późnej( od 20 lat):
- bezsenność lub zwiększona senność;
- częste bóle głowy;
- osłabienie mięśni i poważna duszność;
- skolioza.
Ta forma choroby jest uważana za najmniej niebezpieczną, a współczynnik umieralności jest tu znacznie niższy niż w przypadku rozwoju choroby w dzieciństwie.

Diagnostyczne etapy wyszukiwania
 Wykrywanie choroby odbywa się w kilku etapach.
Wykrywanie choroby odbywa się w kilku etapach.
Najpierw zostały zastosowane główne metody diagnostyczne, a następnie można przypisać dodatkowe badania w zależności od rodzaju i stadium choroby.
Główne metody wykrywania glikogenozy typu 2 obejmują:
- anamneza;
- badania laboratoryjne surowicy krwi;
- ocena objawów klinicznych;
- MRI mięśni kończyn dolnych i innych części ciała;
- RTG klatki piersiowej EKG i EchoCG.
Po przeprowadzeniu głównych czynności diagnostycznych, przypisano następujące: 
- Badanie DNA pod kątem wad genetycznych;
- biopsja i analiza mikroskopowa tkanki mięśniowej;
- badanie krwi w celu określenia poziomu aktywności alfa-glukozydazy.
Jeśli chodzi o późną postać choroby, zastosuj następujące metody: polisomnografia
- ;
- określenie pojemności płuc;
- elektromiografia mięśni szkieletowych.
Na podstawie wyników przeprowadzonych badań i oceny ogólnego stanu pacjenta zaleca się leczenie.
Co zrobić, gdy diagnoza zostanie postawiona
Tak więc, diagnoza jest rozczarowująca i nie ma wątpliwości. Co dalej? W takim przypadku należy postępować zgodnie z następującym algorytmem działań:
- przygotować pakiet następujących dokumentów: wyciąg z karty medycznej, który powinien zawierać informacje o diagnozie, zalecanych lekach i sposobie ich wprowadzenia;protokoły komisji lekarskich;skierowanie na niepełnosprawność;
- przekazać wskazaną listę lekarzowi prowadzącemu w celu przygotowania wniosku o leczenie;
- odwiedzają władze sanitarne wraz z wnioskiem o zakup niezbędnych leków;
- napisać wniosek do organizacji publicznej o pomoc;
- , jeśli wniosek został odrzucony, konieczne jest powiadomienie Ministerstwa Zdrowia i uzyskanie pomocy prawnej.
Jaka współczesna medycyna może zaoferować
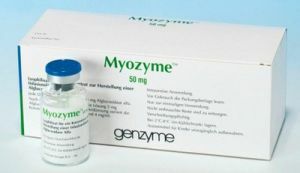 Jak wspomniano powyżej, niemożliwe jest wyleczenie choroby Pompego. Jednak nauka zajmuje się tym problemem od kilku lat i osiągnięto pewne osiągnięcia. Dzisiaj możesz spowolnić proces i złagodzić stan pacjenta. Co oferuje współczesna medycyna w Rosji?
Jak wspomniano powyżej, niemożliwe jest wyleczenie choroby Pompego. Jednak nauka zajmuje się tym problemem od kilku lat i osiągnięto pewne osiągnięcia. Dzisiaj możesz spowolnić proces i złagodzić stan pacjenta. Co oferuje współczesna medycyna w Rosji?
Jedynym lekiem dostępnym dla obywateli rosyjskich jest Mayozaim do podawania dożylnego. Jest to środek na całe życie, którego działanie ma na celu przywrócenie funkcji narządów i układów, na które choroba miała wpływ.
Lek można stosować w każdym wieku i we wszystkich postaciach choroby. Następujące pozytywne efekty odnotowano w wyniku jego stosowania:
- poprawia stan kości i mięśni;
- stabilizuje pracę układu oddechowego;
- zwiększa aktywność ruchową.
Zastosowanie preparatu Mayozeim zmniejsza ryzyko śmierci o 99% i poprawia pracę układu oddechowego o 92%.Jednak u niektórych pacjentów można zaobserwować następujące działania niepożądane leku:
- wymioty i nudności;
- objawy alergiczne na skórze;
- duszność i tachykardia.
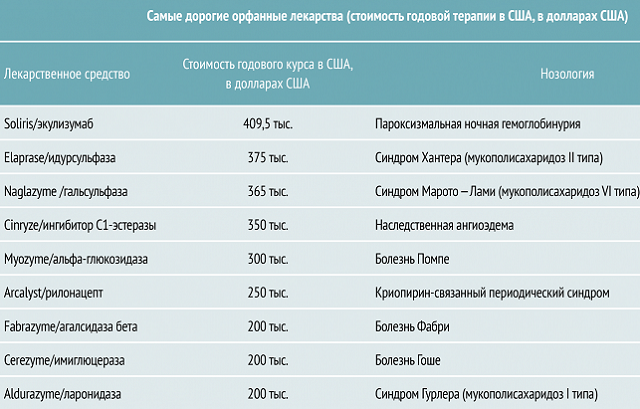
Wadą leku jest jego wysoka cena. Jeśli nie ma możliwości przeprowadzenia leczenia, lekarze korygują stan pacjenta za pomocą tańszych i mniej skutecznych leków.
Rokowanie i zapobieganie
Rokowanie w chorobie Pompego zależy od postaci choroby. We wczesnym stadium infantylnym dzieci umierają, zanim osiągną wiek jednego roku. Przyczyną śmierci jest niewydolność serca.
Późna postać niemowlęca i młodzieńcza pozwoli na osiągnięcie wieku 20 lat.Śmierć pojawia się w wyniku niewydolności krążeniowo-oddechowej.
Najkorzystniejsze rokowanie dla osób, u których choroba została zdiagnozowana po 20 latach. Ci ludzie mogą żyć do późna w podeszłym wieku, ale prawie wszyscy są niepełnosprawni.
Jeśli chodzi o zapobieganie, nie istnieje. Jest to choroba genetyczna i nie ma gwarancji, że zdrowe dziecko urodzi się w rodzinie, w której były podobne przypadki choroby. Możesz jedynie określić stopień prawdopodobieństwa urodzenia chorego dziecka, przechodząc odpowiednie badanie.