 Em 2001, esta síndrome foi incluída na lista de doenças para as quais a atividade epiléptica e os transtornos epileptiformes na eletroencefalografia são características.
Em 2001, esta síndrome foi incluída na lista de doenças para as quais a atividade epiléptica e os transtornos epileptiformes na eletroencefalografia são características.
Estes distúrbios causam deterioração do cérebro em desenvolvimento. Além disso, a hipótese de Otahara foi adotada que esta doença em 80% das situações se desenvolve em outra síndrome de Vest. E depois de algum tempo houve situações com a transformação da doença na síndrome de Lennox-Gastaut.
A síndrome de Otahar é a fase inicial da encefalopatia epiléptica, que é detectada em crianças nos primeiros 3 meses após o nascimento. As convulsões agudas são características da doença, que aparecem ao longo da vida de 10 dias da criança, em casos raros após o nascimento.
Infracções no metabolismo do bebê falam sobre doenças familiares.É exacerbado no contexto da saúde ideal.
Causas e etiologia do
O pré-requisito mais popular é a formação de defeitos cerebrais - megalencephalus unilateral, portentsefalia e outros.Às vezes, como uma fonte pode atuar como uma violação de mapeamento, falhas associadas à troca de substâncias.
Em estudos pessoais, Otahar considerou 10 situações. Como resultado, 2 tiveram um cisto nos hemisférios cerebrais - portentsefalia,  também em 2 síndromes de Aikardi e encefalopatia mista subaguda - uma mudança distrófica no tecido cerebral, o que causa distúrbios nas suas funções. Nos outros 6 filhos, a origem da doença não foi revelada.
também em 2 síndromes de Aikardi e encefalopatia mista subaguda - uma mudança distrófica no tecido cerebral, o que causa distúrbios nas suas funções. Nos outros 6 filhos, a origem da doença não foi revelada.
Em outra pesquisa de 11 crianças, a primeira teve asfixia no nascimento, na 1ª com patologia congênita, um valor importante no desenvolvimento e distribuição da qual é reproduzida pela genética( agenesia do corpo caloso).
Na primeira hiperglicinemia não cetona, as outras bases da doença não são encontradas. Apenas 1 estava com sintomas semelhantes de epilepsia como a de seus parentes.
Schlumberger em seu experimento em 8 de 8 situações diagnosticadas com defeitos do cérebro.6 dos quais - megalencefalia unilateral e uma síndrome de Aikardi. A megalencefalia unilateral como causa foi encontrada no trabalho de pesquisa de Martin, Ohtsuka.
Em 1995, em um artigo dedicado à epilepsia infantil, foi escrito que as fontes da síndrome de Otahar são malformações cerebrais.

Como resultado, após uma multiplicidade de estudos, geralmente foi acordado que distúrbios estruturais dos hemisférios cerebrais são provocadores da doença.
Fisiopatologia
As síndromes de Otahara, Vesta e Lennox-Gastaut são muito estreitamente relacionadas, é aceito que essas síndromes mostram as reações relacionadas ao envelhecimento do cérebro em diferentes períodos de sua formação.
Com este tipo de epilepsia, foi necessário enfrentar outro paciente com malformações hemisféricas ou focais, que produzem convulsões parciais severas que precedem, mas a maioria segue imediatamente após a síndrome de Otahar.
Manifestações clínicas de
Em 2002, Aicardi e Ohtahara apresentaram as seguintes características principais da doença de Otahar:
- geralmente é infectado com lactentes imediatamente após o nascimento ou aos 10 dias de idade;
- diferentes tipos de convulsões, o principal é um espasmo excitatório, convulsões com tensão muscular excessiva podem manifestar-se tanto durante o dia como da noite;
- grave desaceleração na formação psicotrópica, muitas vezes termina na morte na infância;
- possível transformação da doença em outras síndromes;
- basicamente, todos os casos estão associados a distúrbios cerebrais.
Para a síndrome de Otahara, há uma deterioração progressiva da saúde com um aumento no número de convulsões, uma desaceleração visível na formação psicomotriz do corpo. Basicamente, os bebês com este diagnóstico permanecem desativados.
A doença é caracterizada por diferentes tipos de convulsões. Em regra, espasmos excitatórios, que podem se espalhar por todo o corpo, são simétricos e lateralizados em relação aos hemisférios cerebrais. O comprimento médio do ataque é, em média, 10 segundos, com um intervalo de 10-15 segundos. Há também uma pequena chance de desenvolver outras convulsões.
Devido à doença, os bebês são menos ativos, a hipotensão se manifesta. Na síndrome de Vesta, a doença pode ser transformada em 2-6 meses de vida de crianças, de acordo com os resultados do estudo, a porcentagem da transição é de cerca de 75%.No futuro, a doença tem todas as chances de crescer na síndrome de Lennox-Gastaut.

Procedimentos de diagnóstico
Neuroimagem é uma combinação de maneiras pelas quais você pode visualizar a estrutura, funções e propriedades bioquímicas do cérebro. Esses métodos são necessários para determinar as causas e propósitos das terapias de tratamento. Em regra, eles mostram desvios significativos da norma e da malformação.
 Se os resultados da neuroimagem forem normais, o rastreio metabólico deve ser realizado.
Se os resultados da neuroimagem forem normais, o rastreio metabólico deve ser realizado.
Os primeiros sintomas da doença são a eletroencefalografia interictal, que é um padrão de supressão de flash com altas descargas paroxísticas de amplitude, que são separadas umas das outras por uma curva plana com uma duração de aproximadamente 18 segundos.
O padrão de "supressão flash" pode ser assimétrico ou predominar em um dos hemisférios do cérebro, e também piorar no momento do sono.
Se o padrão de "supressão de flash" for substituído em locais por gypsarhythmy( atividade caótica anormal) nos 3-5 meses, isso indica uma doença diferente - síndrome de Vest.
Se a atividade lenta da onda de espiga, é típica da síndrome de Lennox-Gastaut. Em todas as outras situações, desenvolve-se em uma epilepsia parcial grave com alta atividade de células nervosas em um dos hemisférios.
Uma vez que a principal causa da doença é dano cerebral, é necessário realizar um teste de neuroimagem. Todas as mudanças estruturais podem ser observadas com MRI e CT.
Com resultados normais de neuroimagem, o diagnóstico do metabolismo é prescrito. Alguns distúrbios no processo de metabolismo do orgasmo podem levar a lesões dos hemisférios cerebrais.
A eficácia da terapia é praticamente zero.
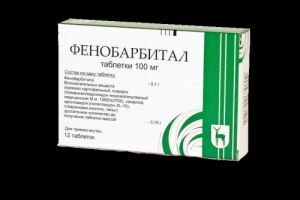 O fármaco antiepiléptico Phenobarbital, também conhecido como Luminal, é capaz de reduzir o número de convulsões, mas os anticonvulsivantes são incapazes de parar o abrandamento da formação psicomotora.
O fármaco antiepiléptico Phenobarbital, também conhecido como Luminal, é capaz de reduzir o número de convulsões, mas os anticonvulsivantes são incapazes de parar o abrandamento da formação psicomotora.
Nenhum dos casos conhecidos teve uma reação positiva ao tratamento com hormônio adrenocorticotrópico e antagonistas de cálcio. Em 2001, Fusco demonstrou terapia favorável com vitamina B6.
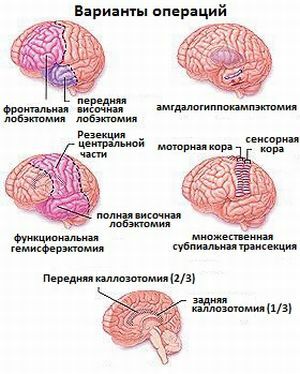
Ohno apresentou uma situação com um bom resultado no modo tratamento com Zonisamida. Em variantes com hemimegalencefalia ou displasia cortical, intervenções neurocirúrgicas podem ajudar.
Infelizmente, atualmente não existe um tratamento farmacológico efetivo da doença, metade dos pacientes morrem desde a infância de várias semanas a um mês, o resto desenvolve um defeito neurológico e psicológico estável.
Muitas vezes, ataques na síndrome de Otahar são irrecuperáveis e não podem ser tratados com drogas antiepilépticas. Com o tempo, a transformação em outras doenças é possível.
Se a transição não foi implementada, o desenvolvimento psicomotor será melhor. No entanto, a previsão é desfavorável, na maioria dos casos um resultado letal foi observado.