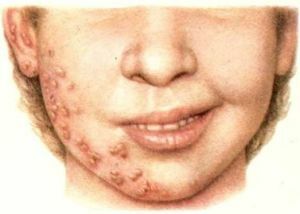
panencefalite é uma forma de encefalite viral. Quando subaguda vantajosamente afetados matéria branca do cérebro. Além disso, há alterações degenerativas em neurónios. Presumivelmente doença alérgica
panencefalite é uma forma de encefalite viral. Quando subaguda vantajosamente afetados matéria branca do cérebro. Além disso, há alterações degenerativas em neurónios. Presumivelmente doença alérgica
tem uma natureza viral. No momento, consideramos a versão sobre a possível origem da doença a partir dos vírus do sarampo, raiva, herpes vírus tipo 3.
três principais formas leykoentsefalita clínica-morfologichekie:
- subaguda esclerosante Van Bogart;
- periaksialny Schilder;
- hemorrágica aguda.
Neste estudo, a encefalite cérebro afetada dá os seguintes resultados: há atrofia do giros eo alargamento sulcos, locais desmielinizantes são encontrados em todas as partes do cérebro, principalmente na substância branca, mas também na substância cinzenta do córtex. Houve uma expansão modesta dos ventrículos.

- fatores precipitantes Conteúdo
- mecanismo de desenvolvimento e
- manifestação sintomas de cada formulário do diagnóstico abordagem
- doença
- para fins terapêuticos previsão
- e mortalidade
- caso clínico verdadeiro
fatores precipitantes
Há especulações de que leykoentsnfalit Van Bogart causada por um vírus do sarampo, que estava emum estado latente nos neurónios do cérebro e activado em certos processos.
esclerose múltipla alternativa em crianças tratadas periaksialny encefalite. Notou a ocorrência frequente de leykoentsefalita hemorrágica aguda após a vacinação.
patogênese da doença é obscura, acredita-se que o vírus é o gatilho no processo auto-imune.mecanismo
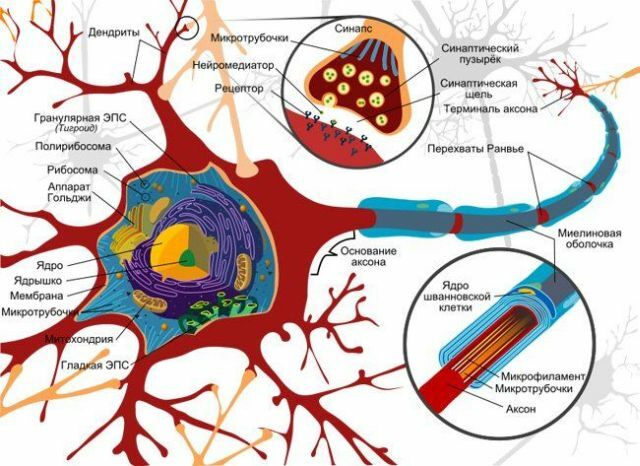
de desenvolvimento e manifestação processo
é mais frequentemente localizadas nos gânglios basais.À medida que a doença progride, as ligações associativas entre o cérebro lóbulos vírus
leykoentsefalita
afectado, definindo desse modo o papel de base de transtornos mentais no quadro clínico.
doença mais comum em crianças, mas houve incidentes de doença em adultos. Geralmente começa depois de uma infecção prévia ou vacinação em pacientes com distúrbios congênitos de imunidade ou neurológicos anormalidades.
primeiros sinais característicos incluem perda de memória, funções corticais, letargia, demência.Às vezes, a doença é acompanhada pelo desenvolvimento de uma síndrome esquizofrênica.perturbações do movimento observadas, convulsões epilépticas.
subaguda evolui rapidamente, com uma média de 1,5-2 anos. Após a ocorrência de morte caracterizada por doenças respiratórias, estado de mal epiléptico.
forma mais comum - leykoentsefality Schilder e van Bogart.síndrome de Van Bogart acompanhada de sintomas extrapiramidais, convulsões epileptiformes, demência.
para a doença de Schilder caracterizada por perturbações do sistema motor de óptica ou nefrite. Os sintomas aparecem
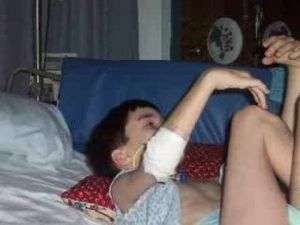 progresso constante, de repente, eles são caracterizados. No início, a doença é acompanhada por transtornos mentais. Em crianças pequenas, isso se manifesta na forma de irritabilidade, apatia, agressividade, letargia.
progresso constante, de repente, eles são caracterizados. No início, a doença é acompanhada por transtornos mentais. Em crianças pequenas, isso se manifesta na forma de irritabilidade, apatia, agressividade, letargia.
gradualmente começam a desaparecer lógica do pensamento, habilidades asseio, vocabulário reduzido. Neste contexto, em poucos meses, começar a desenvolver sintomas neurológicos.
Pode envolver alucinações, aumento do tónus do músculo, agnosia, apraxia, hipercinesia polimórfica. Depois de juntar as alterações do sistema de motor: paresia e paralisia, marcha instável, perda de coordenação.
sintoma constante é uma desordem convulsiva, depois de se juntar distúrbios tróficos e vegetativas. Os sintomas
cada formulário doenças
Quando doença Van Bograrta mais frequentemente do que em outras formas, não é a destruição da medula espinal e do tronco cerebral. Para esta forma leykoentsefalita manifestação precoce característica de distúrbios extrapiramidais, sinais piramidais estão se unindo numa fase posterior. Nesta forma da doença são raros ataques epilépticos.
Na leucoencefalite de Schilder, em contraste com outras formas clínicas e esclerose múltipla, observa-se distrofia precoce dos axônios. Com esta forma prevalecem os sintomas piramidais, acompanhada de convulsões epilépticas freqüentes.
Com encefalite hemorrágica aguda, o aparecimento foi agudo, com aumento de iluminação nos sintomas. Eles estão doentes em 20-40 anos com uma duração de até 2 semanas.É acompanhada por uma dor de cabeça, uma violação da consciência, rigidez dos músculos do pescoço, coma.
Esta forma de leucoencefalite é caracterizada por convulsões e distúrbios do aparelho motor, paralisia. Em casos muito raros, uma forma subaguda ou crônica pode se desenvolver.
Abordagem ao diagnóstico de
Em um estágio inicial de desenvolvimento, a doença é facilmente confundida com esquizofrenia, histeria e neurastenia. Em seguida, ocorre um diagnóstico diferencial com tumores cerebrais 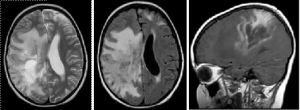 .
.
A natureza difusa da lesão, presença ou ausência de aumento da pressão intracraniana, sinais de deslocamento das estruturas medianas do cérebro no EEG são levadas em consideração. Para confirmar o diagnóstico, CT e estudos imunológicos são realizados.
Estudos de laboratório têm pinocitose moderada no líquido cefalorraquidiano, aumentando os níveis de gama de globulina e proteína. Mudanças na reação de Lange são observadas. O EEG registra os complexos de Rademekker. Os mais informativos são CT e MRI.
Os estágios iniciais da encefalite esclerosante aguda são semelhantes à esquizofrenia. Neste momento, é difícil isolá-lo, mas há uma série de sintomas que ainda permitem excluir a esquizofrenia: perda de memória, distúrbios da fala, exaustão rápida na atividade intelectual.
Nos dados laboratoriais, observa-se a manutenção elevada da fibra e da gama-globulina.
Para a terapia de
 São utilizados medicamentos para corticosteróides. Prednisolona 1-1,5 mg / kg de peso corporal, durante 2-3 semanas, diminuindo gradualmente a dose. Se a terapia dar um resultado positivo, eu continuo em doses de manutenção. Aumento de episódios de exacerbação da doença.
São utilizados medicamentos para corticosteróides. Prednisolona 1-1,5 mg / kg de peso corporal, durante 2-3 semanas, diminuindo gradualmente a dose. Se a terapia dar um resultado positivo, eu continuo em doses de manutenção. Aumento de episódios de exacerbação da doença.
Em paralelo, os antibióticos são prescritos: Penicilina, Eritromicina, Tetraciclina.
Aumentar a ingestão de sais de potássio, enquanto diminui a quantidade de sais de sódio. Se necessário, prescreva vitaminas, bem como drogas psicotrópicas.
Prognóstico e mortalidade
A doença progride de forma constante, sempre levando à morte. A duração da doença é de 0,5 a 3 anos.
Em alguns casos, é possível mais tempo, enquanto a doença prossegue cronicamente, as remissões são observadas periodicamente. Durante a remissão, os sintomas da doença podem estar completamente ausentes.
O verdadeiro caso clínico
Paciente Vadik, 10 anos, entrou no hospital com queixas sobre escolaridade insuficiente e fraco progresso. Hereditariedade: fobia na mãe, o pai é saudável. 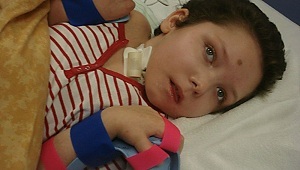
A irmã do pai cometeu suicídio. Na família do pai, há um parente mentalmente retardado. Nascidos da terceira gravidez, os dois primeiros terminaram com trabalho antes do termo, as crianças não sobreviveram.
A terceira gravidez foi desmaie, nasceu durante. O paciente desenvolveu-se sem desvios, visitou o jardim de infância, muitas vezes doente. Em 10 anos comecei a sofrer de enurese. Pouco tempo depois, surgiram medos infundados, o progresso deteriorou-se. Ele começou a ficar atrasado em desenvolvimento, fazendo caretas, tornou-se muito desconsiderado.
Resultados da pesquisa: a pele está seca, avermelhada, o turgor é abaixado, ele se alimenta satisfatoriamente. Há uma pigmentação melhorada no tronco. Periodicamente, diarréia, colesterol 303 mg%, proteína no licor 0,67.Um resultado positivo da reação de Nonne-Apelt.
Estado mental da criança: normalmente suporta a conversa, um rico conjunto de palavras, depois de um tempo começa a acenar os braços, girar o cabelo, especialmente para dobrar o dedo. Rapidamente fica cansado, não se concentra, há dores de cabeça.
Conclusão do endocrinologista: Não há sinais de disfunção do córtex adrenal.
A conclusão de uma neurocirurgião: síndrome de Kerning bilateral, a limitação do olhar para cima, é possível um tumor no lobo frontal.
 Resultados da pneumoencefalografia: a disposição dos ventrículos é simétrica, a ausência de tumor nos hemisférios cerebrais. Hidrocefalia interna com alterações nas meninges.
Resultados da pneumoencefalografia: a disposição dos ventrículos é simétrica, a ausência de tumor nos hemisférios cerebrais. Hidrocefalia interna com alterações nas meninges.
com Penicilina e Biyohinol. Depois disso, o menino foi de alta com um diagnóstico: um processo pouco claro nos lobos frontais.
Após alguns meses, voltei a entrar no tratamento.
Na admissão: desordem da convergência do olho, achatamento da dobra nasolabial esquerda. Diminuição dos reflexos do cremaster e abdominal. A presença de proteína no licor 1,32.
Na inspeção: violações bruscas da mentalidade, fadiga intelectual rápida. Com deficiência, mas manteve as habilidades de leitura e escrita. Há uma segurança na data com os pais. O diagnóstico é leucoencefalite.
No futuro, a doença progride desde o início da doença até o aparecimento de crises epilépticas após 24 meses. Após a hospitalização um ano depois, ele parou de se levantar.Últimos meses completamente desamparados, choraram lindamente. Ele morreu na queda da atividade cardíaca.