İçerik
- Hastalık hakkında genel bilgi
- Kraniostenozun nedenleri ve patogenezi
- Kraniostenoz neden tehlikelidir?
- Hangi doktora gitmeliyim?
- sınıflandırma
- Belirtiler
- teşhis
- Tedavi
- Tahmin etmek
- Kraniostenoz hakkında video
Kraniostenoz patolojik bir durumdur.Kafatasının yarısını birbirine bağlayan metopik sütürlerin erken büyümesi nedeniyle. Sonuç olarak, çocuğun alnında, yüz özelliklerini bozan ve ciddi klinik semptomlara yol açan bir deformasyon şişkinliği oluşur.
Hastalık hakkında genel bilgi
Kafatası, anatomik dikişlerle birbirine bağlanan yüz ve beyin bölümlerinden oluşur.
Beyin yapılarını içeren serebral bölge, parietal ve temporal dahil olmak üzere 4 çift olmak üzere 8 kemikten ve benzer sayıda tek kemikten oluşur:
- önden;
- oksipital;
- kama şeklinde;
- kafes.
Katı yapıların temas hatlarına kafatası sütürleri denir. Embriyonik ve doğum sonrası dönemlerde, 3 gelişim aşamasından geçerler - membranöz, kıkırdaklı, kemik. Kürlenme süreci 25 yaşına kadar tamamlanır.
Yenidoğanlarda, sagital sütür yaşamın ilk yılı boyunca açık kalır. Çeşitli etiyolojilerdeki patolojik değişikliklerle, çok hızlı büyür, bu da sert liflerin normal büyümesine müdahale eder.
Anomaliye, kraniyal kemiklerin, beyin dokularının, sinir liflerinin gelişiminin ihlali eşlik eder. Kraniostenoz, sürekli olarak yüksek tansiyon ve hemodinamik bozukluklarla karakterizedir.
Optik sinirleri etkileyen patoloji, zihinsel gelişimde gözle görülür bir gecikmeye yol açar. Dünyadaki çeşitli anatomik deformite formlarının prevalansı, toplam yenidoğan sayısının% 0.03-3.5'i düzeyinde kaydedilir.
Erkek çocuğun alnında metopik dikiş, kız çocuğuna göre daha sık görülür. Pediatrik uygulamada en yaygın olanı, erken yaşta anatomik anomalilerin monosinostotik formudur.
Skafosefali adı verilen sagital sütürlerin erken füzyonu, klinik olarak bildirilen kraniostenozların yaklaşık %50-65'inden sorumludur. En nadir ve prognostik olarak elverişsiz patolojik bozukluk tipi sendromiktir.
Şiddetli klinik semptomların kapsamlı bir kompleksi eşlik eder. Bu tür kraniyostenoz, yaşamın ilk 10-12 ayında artan bebek ölümü olasılığı ile karakterize edilir. Kraniyal boşluğun yetersiz genişlemesi beyinde mekanik ve kompresyon hasarına yol açar.
Kraniostenozun nedenleri ve patogenezi
Sagital sütürdeki erken aşırı büyümenin etiyolojik faktörleri güvenilir bir şekilde belirlenmemiştir.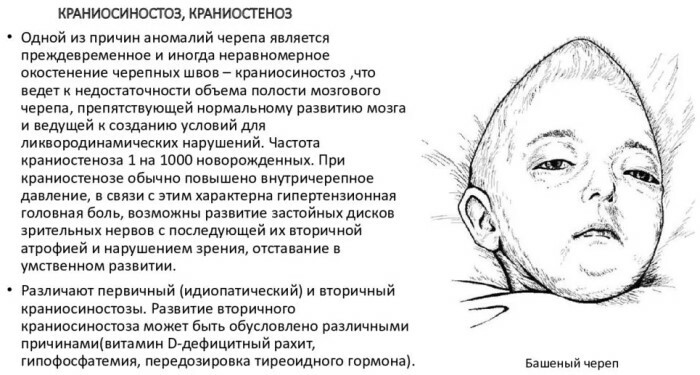
Muhtemelen, kraniyostenoz aşağıdakiler tarafından provoke edilir:
- intrauterin gelişim sırasında hormonal bozukluklar;
- perinatal hasar;
- annenin hamilelik sırasında yaşadığı stres;
- rahim boşluğunun dar iç boşluğu ile embriyonun kafa kemiklerini sıkmak;
- genetik patolojiler;
- kromozomal mutasyonlar;
- radyasyona maruz kalma;
- kanserojen etkiler;
- teratojenik özelliklere sahip yasaklanmış ilaçların bir çocuğunu taşırken bir kadın tarafından alınması;
- fetüsün sinir sistemine hipoksik hasar;
- annenin gebelik dönemindeki bakteriyel-viral veya mantar hastalıkları;
- prematürelik.
Sunulan nedenler tamamen varsayımsaldır ve hiçbir bilimsel temeli veya kanıt temeli yoktur. Muhtemelen Aper sendromu metopik sütür oluşumunda rol oynar.
Bu genetik patoloji, kraniyal kemikleşme sürecini bozar. Aynı olasılık derecesinde, kraniyostenoz, nadir bir kalıtsal hastalık olan Cruson sendromunun arka planında ortaya çıkar.
Sapma, kranyumun yüz ve beyin loblarının ilerleyici deformitesi ile klinik olarak ifade edilir. Araştırmacılar, sagital dikişin erken aşırı büyümesinin genetik olarak belirlenmiş bir başka olası nedeni, Pfeiffer sendromu diyorlar.
Kalıtsal doğanın ihlali, baskın otozomal iletim mekanizmasını karakterize eder. Hastalık, uzuvlar ve kafatası dahil olmak üzere iskelet yapılarının oluşumunu bozar.
Son çalışmalar, fibroplastik hücrelerin büyüme faktörünün reseptör elemanlarının oluşumundan sorumlu bir gen mutasyonunun kraniyostenozunun gelişimindeki öncü rolü nispeten doğru bir şekilde belirlemiştir.
Patogenetik olarak, anomaliye bir veya daha fazla kemik ekleminin erken sinostozu neden olur. Sagital sütüre ek olarak, koroner ve lambdoid çizgiler erken füzyona karşı hassastır.
İhlal, kemik dokusunun dik olarak yönlendirilmiş telafi edici büyümesinin etkisine neden olur. orijinalin bölünmesinin bir sonucu olarak yeni hücresel elementlerin oluşumunu doğrulayan Rudolf Virchow yasası yapılar.
Bir çocuğun alnındaki metopik dikiş, anormal derecede hızlı proliferatif sürecin bir sonucu olarak ortaya çıkar ve bu da kraniyal deformitelere neden olur. Patogenetik özellikler, füzyon zamanı, inert yapıların büyüme hızı, zamanından önce kapanan eklemlerin sayısı ve konumu ile belirlenir.
Kraniostenoz neden tehlikelidir?
Anomaliye, nörolojik semptomlar, fundusun venöz tıkanıklığı, ışığa duyarlı optik liflerin kısmi veya tam atrofisi ile kendini gösteren hipertansif bir etki eşlik eder.
Kraniostenoz, çok sayıda olumsuz sonuç, sistemik patolojiler ve ciddi komplikasyonlar ile karakterizedir. Anatomik anomalinin patogenetik resmine bağlıdırlar.
Kraniostenozun en sık görülen komplikasyonları şunlardır:
- spinal fıtık çıkıntıları;
- kafatasının ön lobunun deformasyon şişkinliği;
- tam körlük olasılığı ile azalmış görme keskinliği;
- entelektüel gelişimde gecikme;
- kozmetik kusurlar;
- şaşılık;
- göz yuvalarının az gelişmişliği;
- değişen yoğunluk ve süreye sahip düzenli sefalik paroksizmler;
- spontan kas krampları;
- epileptik patolojinin gelişimi;
- kalıcı kas-iskelet bozukluklarına yol açan eklem disfonksiyonları;
- hidrosefalik sendrom;
- uyku apnesi;
- solunum bozuklukları.
Sonuçların doğası ve ciddiyeti, kraniyostenoz şekli ile belirlenir. Sagital sütürün erken füzyonu olan yaklaşık her 5 çocukta, yüksek sinir aktivitesinin ciddi bozuklukları ile desteklenen intrakraniyal hipertansiyon vardır.
Kraniostenoz için oftalmik patolojiler, servikal omurgadaki tıkanıklık tipiktir. Uluslararası araştırmalara göre, metopik sütür oluşumu vakalarının %50-60'ında çocuğun bilişsel yeteneklerinde azalma gözlenmektedir.
Kraniyostenozun yaygın bir komplikasyonu, göz kürelerinin anatomik yatağın dışında antero-yüz yönünde tek veya binoküler yer değiştirmesi olan ekzoftalmidir.
Konjenital bir bozukluk tehlikesi düzenli konvülsif nöbetler, şiddetli baş ağrısı, mide bulantısı ve kusmadır. Bu tür çocukların bilgileri ezberleme ve öğrenme konusunda keskin bir yeteneği vardır.
Hangi doktora gitmeliyim?
Kraniyostenozun tedavisi, anomalinin neden olduğu eşlik eden semptomlar ve komplikasyonlar, bebeğin hastanede ilk muayenesini yapan bir neonatolog tarafından gerçekleştirilir. Gelecekte, hasta bir bölge çocuk doktoru tarafından denetlenir.
Hastalığın özgüllüğü ve patogenezi dikkate alındığında, maksillofasiyal ve pediatrik beyin cerrahlarının müdahalesi olmadan yapılamaz. Çocuk doktoru, deformasyon değişikliklerinin ameliyatla düzeltilmesine hazırlanmak için ebeveynleri bu profilin uzmanı olan bir çocuğa danışmaya yönlendirir.
sınıflandırma
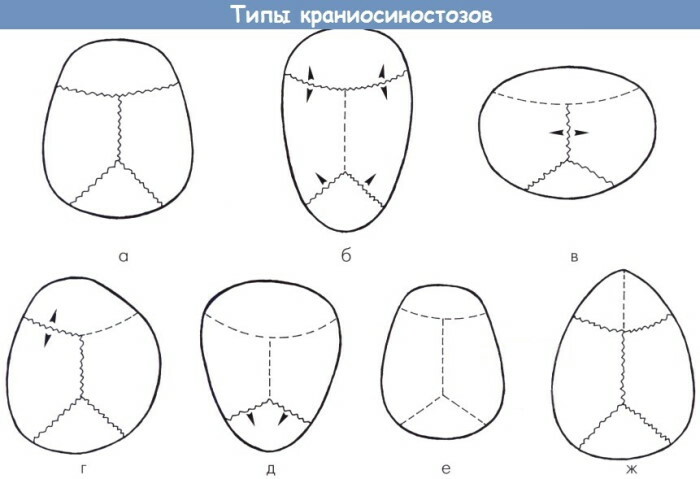
Kraniostenoz, gözlenen klinik tabloya, iddia edilen etiyolojik faktörlere ve erken kaynaşmış kraniyal eklemlerin sayısına göre 5 forma ayrılmıştır.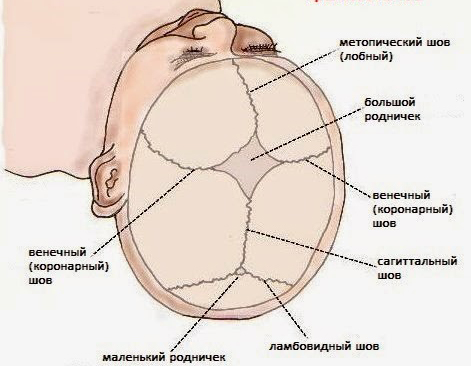
Konjenital anomalilerin mevcut sınıflandırması tabloda sunulmaktadır:
| Çeşitlilik | Patogenetik ve klinik özellikler |
| sendromik | Anatomik bozukluğun en şiddetli şekli. Sendromik patoloji sayısız intrauterin malformasyonla birleştirilir. Bu sınıflandırma kategorisi, X kromozomu, monogenik ve diğer kraniostenozların bağlantısını içerir. Bu form, yüz yüzeyinin kemiklerinin displazisi ile sütürlerin erken füzyonunun bir kombinasyonu ile karakterize edilir. |
| Yalıtılmış | Patoloji bağımsız olarak ortaya çıkar ve eşlik eden patolojilere bağlı değildir. Çoğu zaman, bu tür kraniyostenoz, ciddi sistemik komplikasyonlar ve kafatasının belirgin anatomik deformitesi olmadan ilerler. |
| monokraniyostenoz | İhlal, bir sütür bağlantısının erken füzyonunun bir sonucu olarak ortaya çıkar. Monokraniyostenozun en yaygın biçimleri koroner ve lambdoid yıkımdır. |
| polikraniyostenoz | Patolojik süreç, ciddi komplikasyonlara ve belirgin nörolojik semptomlara neden olan 2-3 bağlantı hattını içerir. |
| pansiokraniostenoz | Anomali, tüm kraniyal sütürlerin erken füzyonu ile karakterizedir. Konjenital anomalinin en nadir formunun cerrahi olarak onarılması zordur. |
Metopik sütür başka sınıflandırma sistemlerine sahiptir. Çocuğun alnında anormal bir şişkinlik oluşumunun iddia edilen nedenine bağlı olarak aşağıdaki kraniostenoz varyantları ayırt edilir:
- doğuştan. Bu patoloji, yalnızca genetik mutasyonlar tarafından provoke edilmeyen intrauterin bozukluklardan kaynaklanmaktadır.
- Edinilen. Sık vakalarda, iskelet kemiklerinin sistemik patolojilerinin arka planına karşı gelişir.
- Fizyolojik. Bu tür kraniyostenozun, kafatasının büyümesi sırasında ortaya çıkan embriyonik nedenleri yoktur.
- Patolojik. Hormonal, endokrin ve diğer faktörlerle ilişkilidir.
- Yapay. Bu tür kraniyostenoz, kraniyal kemiklerin büyük ölçekli yapısal kusurlarını ortadan kaldırmak için özel olarak cerrahi bir yöntemle oluşturulur. Bir sonraki ameliyatta deformite düzeltilir.
Kraniostenoz travma sonrasıdır. Bu anormal bozukluk, doğum sırasında kafatasının hasar görmesi veya yenidoğanın deforme edici etkilerinin bir sonucu olarak ortaya çıkar.
Belirtiler
Yenidoğanın ilk muayenesinde patolojik anormalliğin klinik belirtileri fark edilir. Tüm kraniyostenoz formları, plajiyosefali - asimetrik bir yapı veya kafatasının kavisli bir durumu ile karakterize edilir.
Normalde 12-18 aylıkken kapanan büyük fontanelin erken enfeksiyonu vardır. Poliostenotik tipte bir hastalık veya hidrosefalik anomali ile 3 yıla kadar açık kalır.
Yenidoğan döneminde, kraniyostenozun klinik belirtileri şunlardır:
- stabil intrakraniyal hipertansiyon;
- nörolojik bozukluklar;
- endişe;
- aktif ağlama;
- kusma ile mide bulantısı;
- uyku fonksiyon bozukluğu;
- iştahsızlık;
- Gref sendromu - beynin ventriküler bölgelerinde aşırı beyin omurilik sıvısı birikmesi;
- sürekli sarsıcı hazırlık.
Her anormal sapma biçimi, yenidoğan döneminde bireysel patojenetik özelliklere sahiptir. Skafosefali adı verilen sagital dikişin erken kapanması, kafatasının skafoid konfigürasyonu ile belirlenir.
Semptomatik olarak, bu kraniyostenoz formu, kafanın patolojik olarak küçük bir genişliği ile kafatasının ön-arka düzleminin artan boyutu ile kendini gösterir.
Görsel incelemede, fark edilir:
- kemik oluşumlarının uzun yapısı;
- temporal lobların depresif durumu;
- ön yüzeyin çıkıntısı ve kafatasının oksipital kısmı;
- oval bir konfigürasyonun kazanılmasıyla ön düzlemin daralması.
Herhangi bir kraniyostenoz türünün tipik bir belirtisi zeka geriliğidir. Lambdoid kavşağın erken enfeksiyonu, oksipital bölgenin gözle görülür bir düzleşmesini karakterize eder.
Bu kraniyostenoz formunda nörolojik semptomlar minimaldir veya yoktur. Koroner anomali tek taraflı veya iki taraflıdır. Birincisi, kafatasının ön düzleminin deformasyon düzleşmesi ile karakterizedir.
Büyüdükçe semptomlar büyür ve kendini gösterir:
- burun köprüsünün eğriliği;
- elmacık kemiklerinin şeklinde bir değişiklik;
- üst çenenin deformasyonu nedeniyle ısırık ihlali;
- şaşı
Bilateral koroner kraniostenozda, yörüngelerin kenarlarının düzleşmesi, ön düzlemin genişlemesi, akrosefali - kafatasının anormal şekilde uzamış "kule benzeri" bir şekli gözlenir.
Nörolojik semptomlar spesifik değildir ve diğer patoloji türleriyle aynıdır. Trigonosefali olarak adlandırılan, topikal olmayan kraniyostenoz formu, kafatasının üçgen bir bölümü ve kemikli bir omurga oluşumu ile karakterizedir.
Bir çocuğun alnındaki metopik sütür, hipertelorizm - yörünge yer değiştirmesi ve yörüngeler arasındaki mesafedeki azalma ile birleştirilir. Yaşlandıkça, kemik tepesi düzleşir, alnın şekli normale yakın bir konfigürasyon kazanır. Trigonosefali semptomatik olarak görme bozuklukları, zihinsel bozukluklar ile kendini gösterir.
Hastalığın en şiddetli sendromik formunda aşağıdakiler gözlenir:
- Solunum yetmezliği;
- yutma zorluğu;
- belirgin oftalmik patoloji;
- şiddetli baş ağrısı;
- aşırı psikomotor ajitasyon.
Kafatasının yüz kısmının kemik yapılarının displazisi, göz kürelerinin çıkıntısı not edilir. Şiddetli ihlaller çocuğun 12 ay içinde ölümüne yol açar. doğumdan sonra.
teşhis
Konjenital bir hastalığın tipi, anormal değişikliklerin derinliği, patogenetik özellikleri birincil fizik muayene, enstrümantal ve laboratuvar yöntemleri ile belirlenir.
Öykü çok bilgilendirici değildir, ancak neonatolog veya çocuk doktorunun yenidoğanın durumundaki dinamik değişiklikleri izlemesine olanak tanır. Laboratuvar testleri belirli belirteçleri tanımlamaz.
Sadece komplikasyonların ciddiyetini teşhis etmek ve eşlik eden patolojileri tespit etmek için kullanılırlar. Kemik deformitelerini görselleştirmeye yönelik donanım yöntemleri, kraniyostenoz için standarttır.
Serebral dokulara verilen hasarın derecesini değerlendirme yöntemleri şunları içerir:
- nörosonografi - kafatasının iç boşluklarının ultrasonik dalgalarla iki boyutlu sektörel taraması;
- X-ışını - kafanın çalışılan bölgesinin görüntüsünün sabitlenmesi ile radyasyon teşhisi;
- araştırılan alanın üç boyutlu bir modelini oluşturmanıza izin veren bilgisayarlı tomografi;
- beyin yapılarının iç yapısını detaylandıran manyetik rezonans görüntüleme.
Nörosonografik teknoloji, hipertansif belirtileri tanımlamak için intrakraniyal dokuların durumunu ve ventriküler boşlukların boyutunu değerlendirmek için kullanılır. X-ışını görüntüsü, kemik deformitelerinin doğasını, kraniyal eklemlerin sertleşme derecesini belirler.
Çocuğun alnındaki metopik sütür mutlaka BT ve manyetik rezonans görüntüleme kullanılarak teşhis edilir. Bu tür prosedürler daha bilgilendirici sonuçlar sağlar.
Ek olarak, görme bozukluğunun derecesini belirlemek için oftalmoskopi yapılır. Kraniyostenozun ayırıcı tanısı, anatomik anomalinin tipini netleştirmek ve bir cerrahi rekonstrüksiyon yöntemi seçmek için yapılır.
Tedavi
Kafatasının doğal şeklini geri kazanmanın ve komplikasyonların şiddetini azaltmanın tek yolu ameliyattır. Cerrahi düzeltme için en uygun zaman 6-9 aylık bir çocuğun yaşı olarak kabul edilir.
Bu dönemler, yaşamın bu döneminde serebral bölgenin doku yapılarının yoğun gelişmesinden kaynaklanmaktadır. Büyümeleri kemik çarpıklıkları tarafından engellenir. Bu yaşta, tam olarak sertleşmemiş dokular, ciddi komplikasyon riski olmadan hızla doğal yapılarını eski haline getirir.
Rekonstrüksiyon kapsamı ve operasyon tekniği, patolojinin tipine, anormal değişikliklerin derecesine göre seçilir. Bir çocuk 2-3 yaşına geldiğinde, sadece küçük kozmetik kusurları ortadan kaldırmak için cerrahi rekonstrüksiyon mümkündür.
Düzeltme, kafatası kasasının kemik oluşumlarının yeniden modellenmesinden oluşur. En iyi sonuç, pediatrik maksillofasiyal ve beyin cerrahı tarafından ortaklaşa yapılan bir operasyonla elde edilir.
Trigonosefali ile, daha sonra, özel bir sabitleme kaskı takmayı içeren düzeltici ortezler reçete edilir. Böyle bir dinamik sistem, yoğun kafa büyümesinin neden olduğu artık deformasyonları ortadan kaldırmak için tasarlanmıştır.
Bir kask-ortezi, bir bebeğin hayatının ilk 6-18 ayında en iyi sonuçları verir. Cerrahi düzeltme ve dinamik bir sabitleme sisteminin kullanımına ek olarak, çocuğun beslenme rejiminde yaş gereksinimlerine ve gözlenen ihlallere göre bir değişiklik gösterilir.
Araya giren patolojilerin ilerlemesi veya nörolojik semptomlarda bir artış ile karmaşık ilaç tedavisi reçete edilir.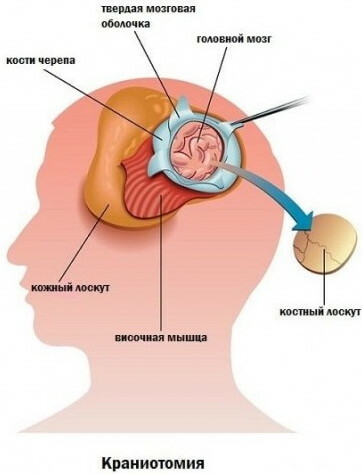
Kranial kemiklerin deformitelerini yeniden yapılandırmak için aşağıdaki kraniotomi türleri kullanılır:
- periostun kısmi amputasyonunu sağlayan lineer;
- 3-5 yaş arası çocuklar için gerçekleştirilen ve intrakraniyal hipertansiyonu ortadan kaldırmayı amaçlayan dolaşım;
- parçalı - kranial kasayı kesmek;
- dekompanse kraniyostenoz formunu düzeltmek için tasarlanmış çift taraflı flep.
Rezeksiyon çizgileri önceden tıbbi bir işaretleyici ile işaretlenir. Tipik etki alanları ön lob, ön fontanel, koroner kavşaktır.
Tahmin etmek
Zamanında ve başarılı bir cerrahi operasyon ile dolu dolu bir yaşam şansı yüksek olarak değerlendirilir. Doğru tahminler, anomalinin şekline, etiyolojik faktöre ve eşlik eden patolojilerin varlığına bağlıdır.
Genellikle metopik sütürleri tamamen ortadan kaldırmak, nörolojik semptomları ve kafa içi hipertansiyonu durdurmak mümkündür. Çocuğun alnındaki patolojik oluşum, yeniden yapılanan kemikler büyüdükçe kaybolur. Sadece sendromik kraniyostenoz prognostik olarak olumsuzdur.
Kraniostenoz hakkında video
Malysheva, bir çocukta kafatasının deformitesi hakkında: