 Gallervordena-Spatz hastalığı( progresif sertliği ) - nadir kalıtsal nörodejeneratif hastalık olan bazal ganglion kaybı ve bunların demir birikmesi vardır.
Gallervordena-Spatz hastalığı( progresif sertliği ) - nadir kalıtsal nörodejeneratif hastalık olan bazal ganglion kaybı ve bunların demir birikmesi vardır.
Birikim işlemleri siyah bir maddede ve soluk bir balonda gerçekleşir. Ayrıca kortekste, değişiklikler sinir hücrelerinde gözlenir, pigmentli ve glial hücrelerin nevrogli görünüm Alzheimer hastalığına benzeyen gliyami.
Hastalık, ilk kez adı Alman araştırmacılar Julius Gallervorden ve Hugo Spatz tarafından tanımlandı.
Bununla birlikte, modern uluslararası tıbbi pratikte hastalığa beyinde demir birikimi ile nörodejenerasyon denir( NBIA).Orijinal ismi reddetmek, Julius Gallervorden'in faşist Almanya'da ötenazi üzerine araştırma yapması gerçeğine bağlı.
Hastalık nadir olarak kabul edilir, 1 milyon kişi için 3 kişiye kadar hastalık söz konusudur. Bu nörodejenerasyon Parkinsonizma, distoni, demans ve ölüme yol açabilir.
genetik hastalık
yetmezliği Gallervordena-Spatz kalıtsal genetik bozukluktur bulunmaktadır. Hastalık hem aile hem de sporadik olabilir( kazara geçer), ancak her zaman kalıtsaldır. Hastalık otozomal resesif kalıtım anlamına gelir.
Hastalığın bulaşması için, her iki ebeveyn de hastalığın heterozigot taşıyıcıları olmalı ve mutasyona uğramış bir allele sahip olmalıdır. Kromozom 20 lokus 20r12.3-P13 bulunan bir gen pantotenat kinaz( PANK2) içinde bir mutasyonun neden olduğu
hastalığı.bu da, amino asit sistein ve pantetein birikimini inhibe kodlayan pantotenat kinaz proteinin 2, sorumlu pantotenat kinaz geni. 
Sonuç proteinleri üzerinde olumsuz bir etkisi vardır ve başlangıç sağlamak demir iyonları ile kimyasal bileşiklerin oluşumu nöronların programlanmış hücre ölümüne yol açar peroksidasyon, işler.Ölü nöronlar yerine glial doku büyür( sinir dokusunun yardımcı hücreleri).
Patolojik süreçler pallid küre, kırmızı çekirdeğin ve siyah maddenin( nigra maddeleri) meydana geldiğini göstermektedir. Demir içeren yeşilimsi kahverengi bir maddenin birikimine neden olurlar. Buna ek olarak, beyin, serebral korteks, periferik sinir gövdeleri ve küre formasyonu neyroaksonalnye görülen omurilik beyaz maddesinde( akson ve boru şeklindeki zar yapılarına büyümesi genişler).
Patoloji formları
Gallervorden-Spatz sendromunda üç form ayırdedilir. Bölüm, hastalığın gelişme yaşına dayanır:
- Pediatrik form .Hastalığın gelişmesinin başlangıcı 10 yıla kadar yaştadır.
- 'nin çocuklu( ergen) formu. Hastalık 10-18 yaş arasında gelişir. 'nin yetişkin( atipik) formu. Hastalık 18 yaşından sonra kendini gösterir. Hastalığın Özellikleri
klinik bulgular her durumda değişebilir ve semptomların tezahürü, bir kişide hastalık şeklinde bağlıdır.
Gallervorden Spat'ın pediatrik formu 5 ila 10 yaş arasında görünür. Hastalığın klasik bir formu olarak kabul edilir.olguda( % 90) ezici sayısı ise
, hastalık bacak kasları etkileyen torsiyon distoni ile başlar. Hastanın kas kasılması, yürüme zorluğu, yürüme değişikliği. Daha sonra yüz kasları, farenkut ve gövde etkilenir.
blefarospazm olası varlığı, ellerin spazmı, spazmodik tortikolis, yüz hemispasm. Hastaların üçte birinde kas sertliği, hipokinezi, tremor( parkinsonizm sendromu belirtileri) var. Hastalığın alt form
tipik belirtileri de içerir:
- epileptik sendromu;
- saldırganlık;
- zihinsel gerilik( zayıf bellek ve dikkatten dolayı);
- asosyalite;
- görme bozuklukları( optik atrofi, retinal dejenerasyon);
- zihinsel bozukluklar. Hastalığın Çocuk
Ergen formu daha yavaş gelişir ve devam eder. Hastalığın başlangıcında kendini gösterecektir: 
- odak torsiyon distoni( genellikle bacaklarda ve çene ağız kaslarının kasları etkiler);
- nöropsikolojik bozukluklar;Davranış bozuklukları
- ;Zihinsel bozukluklar
- .
atipik hastalığı formu - en sık( vakaların% 15) ve ilk iki formlarından farklı ilerler. En tipik belirtileri, bu formu için: bir ayakta durma konumunda dengesini korumak için yetersizlik ile
- Parkinson sendromunun;gövde, distoni, atetoz ve kore( kesik kesik ve yavaş hareket), hemiballismusu( süpürme hareket), miyoklonus( uzatılmadığı kas krampları) farklı bölgelerinde
- istemsiz hareketler;
- demans;
- epileptik sendrom;
- Depresyon, saldırganlık;nedeniyle beyin nöronları yıkımı meydana
- patolojik refleksler.
Tanı kriterleri ve yöntemlerine
Tanı "Gallervordena-Spatz hastalığı" nedeniyle polimorfizmi belirtileri nörologlar için sorunlu olabilir.
Tanı için ana ölçütler:
- hastalığın başlangıcı 30 yıla kadar;
- MRG sonuçlarında tipik resim;
- semptomların sürekli gelişimi;
- ekstrapiramidal sendromlar;
- piramit işaretleri;
- epileptik nöbetler;
- bilişsel bozukluk;
- retinal pigmentez dejenerasyonu;
- aile öyküsünde hastalığın varlığı.Huntington hastalığı, Wilson hastalığı, horeoakantotsitozom, Machado-Joseph hastalığı: - kontrastlı Kaplan" MR
« Göz hastalığı Gallervordena
Spatz hastalığında tipik bir model benzer bir klinik tablo olan diğer hastalıklardan ayırt edilmelidir.
hasta Bu anketlerin yöneliktir teşhis için: Nöropatoloji
- Çalışması nörolojik durumu.
- Elektroensefalografi .
- Göz doktorunun danışması .Direkt oftalmoskopi, görme keskinliği kontrolü yapmak mümkündür.
- Genetik çalışmalar , devralma türünü belirlemek için.
- Araştırma DNA ( genindeki PANK2 tespit mutasyonlar).
- Beynin Pozitron Emisyon Tomografisi .Tespit ön-yan loblar striatum globus pallidus kan akışını azaltmak için gerçekleştirilir.
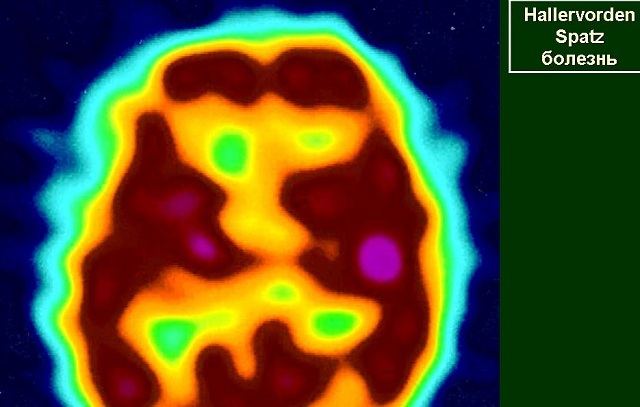
- Beynin manyetik rezonans görüntülemesi .nedeniyle demir birikimine oluşur hipointenstir alanında hiperintense oval kısmı - "kaplan göz" tespit etmek için gereklidir.gelişme "Kaplan Gözü" nin tek teori yoktur, hastalığın klinik belirtilerinin başlaması önce onun görünümünü ve hastalığın başlangıcından sonra yıl içinde görünüşü hakkında spekülasyon var. Ayrıca MR, sfero nöro-aksiyel oluşumları tespit etmeyi sağlar.
Modern doktorlar nasıl yardımcı olabilir?
Modern tıpta, hastalığın önlenmesi veya Gallervordena-Spatz durduramaz hiçbir tedavi yoktur.
Tedavi yardım ve yoksunluk belirtileri yoğunluğu yöneliktir:
- Parkinson sendromu uygulanan dopamin agonistleri ( pronoran, Pramipeksol, Mirapex, piribedil), amantadin
 ( Symmetrel, Midantan).Bununla birlikte, sendromun tedaviye direnci sıklıkla görülür.ödem
( Symmetrel, Midantan).Bununla birlikte, sendromun tedaviye direnci sıklıkla görülür.ödem - hiperkinezi kullanımı valproat ( Konvuleks, Depakinum, Enkorat), benzodiazepinler ( klonazepam, diazepam) için.
- ( Mydocalm, baklofen) kullanılan spastisite, kas gevşeticiler kaldırın.
- Epilepsi nöbetleri, Tomapaksom çıkarılan valproat vardır.
- kognitif bozukluk Gliatilin, Neuromidin uygularken. Ruhsal bozuklukların tedavisi için
- alımı nöroleptikler önerilir( klonazepam, Ketiapin, Rispolenta), antidepresanlar ( Dapoxetine, Sitalopram, venlafaksin).
Hastalığın tedavisinde yeni yöntemler ortaya çıkıyor. Bu pantotenik asit, beyin( globus pallidus) manyetik uyarım uygulanmasıyla tedavisini içerir.
hastalık Gallervordena-Spatz belirtiler sürekli ilerliyor üzücü ama umutsuz değil
.En zoru, çocuğun hastalığın şeklidir.10-15 yıl klinik belirtilerin ilk ortaya çıktıktan sonra bireysel sakatlık takımları.
Hastalığın en olumlu gelişimi hastalığın yetişkin formunda öngörülmüştür.Özellikle hafif bunama durumunda.
Terapi göreli hastanın yaşam kalitesini ve self-servis yeteneğini saklamanızı sağlar. Hastalığın atipik formda beklenen yaşam süresi Gallervordena-Spatz 20 yıldan fazla olabilmektedir.