Günümüzde modern tıp işlemek mümkün değildir yetim( nadir) hastalık vardır.fırsat sadece orada yaşam kalitesini geliştirerek bir ölçüde bu hastalara destekleyecek ama hastalık imkansızdır yenmektir. Bu hastalığın tedavisi için yollar, bilim henüz bulamamıştır rağmen
Günümüzde modern tıp işlemek mümkün değildir yetim( nadir) hastalık vardır.fırsat sadece orada yaşam kalitesini geliştirerek bir ölçüde bu hastalara destekleyecek ama hastalık imkansızdır yenmektir. Bu hastalığın tedavisi için yollar, bilim henüz bulamamıştır rağmen
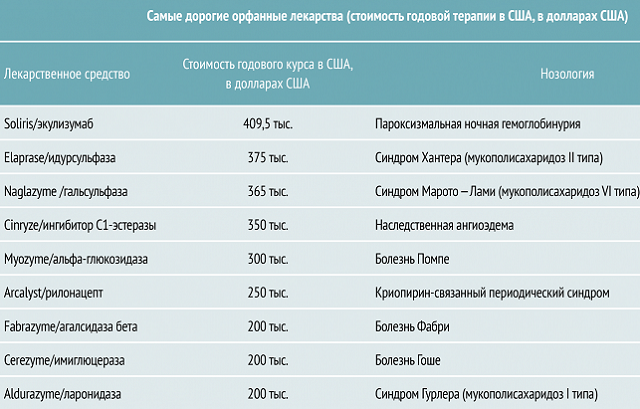
Pompe hastalığı( jeneralize glıkojenez) şu anda, bu hastalıkların listesine ait değil. Ancak, Rus hastalar devletten yardım, kamu kuruluşları ve vakıflar almak kendi ülkesinde ve belirli koşullar altında tedavi edilmesi için fırsat var.
İçerik
- genel özellikleri
- Tarihi ve İstatistikleri
- neden olur ve geliştirme
- Kalıtım
- Modern sınıflandırma
- belirtilerin mekanizması ve klinik yaş
- bağlı modern tıp
- tahminlerini ve
önleme sunabilir Ne tanısı ne yapmalı rapor arama AşamalarıGenel özellikler
Pompe sendromu gövdenin kas ve sinir hücreleri etkiler. Bu durum, genetik seviyede mutasyon değişikliklerin bir sonucu olan alfa-glikozit eksikliği olur.
hasta doğru tedaviyi, artık normal şekilde çalışmaya yapabiliyor bundan sonra kas dokusu yapısının değişiklikleri almazsa. Zamanla, işlem ilerledikçe, hasar şiddetli ağrı eşlik eder diğer kas gruplarına, yayılır.Şiddetli aşamalarda
hastalar mekanik havalandırma için makinalara bağımlı hale gelebilir solunum sisteminde bağımsız ve kas lezyonları pahasına hareket yeteneğini kaybeder. Geçen yüzyılın 30s -

Tarihi ve İstatistikleri
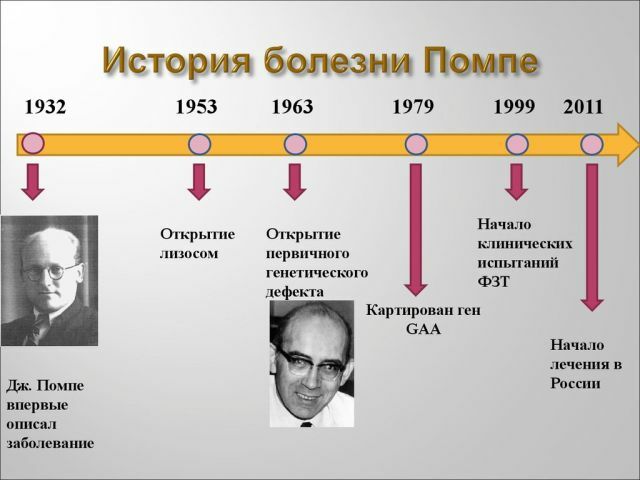
hastalığı nispeten son zamanlarda ortaya çıkardı.İlk kez Hollanda, I.Pompe'den bir bilim adamı tarafından tanımlandı.Her iki cinsiyetten eşit olasılıkla vurmak mümkün
hastalığı, bu ağırlıklı olarak erkek veya kadın değildir. Hastalığın erken formu doğmamış çocuk ve yetişkin Pompe hastalığına 140,000 1 60,000 insanları etkileyen 1 oluşur.2006 yılında
, metodoloji tür hastaları desteklemek için izin verir ve 2013 yılında etkili yollar bulmaya ve Rus bilim adamları ABD'de geliştirilmiştir. Ne yazık ki, tedavi maliyeti çok yüksek olduğundan, her hasta tedavi edilemez.
neden olur ve geliştirme
hastalığın mekanizması iskelet kası ve miyokard içinde mevduat glikojen nedeniyle gelişir. Bu işlem, GAA genindeki mutasyonlar nedeniyle bağlı olan etkilenen kas, kalp kası, sinir sistemi ve karaciğere.İşlem ilerledikçe, ve distrofik dahil olmak üzere zarar, çeşitli hücrelerde meydana gelir.
Pompe hastalığı genelleşmiş ve kaslı.İlk durumda, aynı zamanda, kas sistemi tarafından etkilenen, karaciğer ve böbrek gibi organların değişiklikler vardır. Buna ek olarak, sık kalp yetmezliği belirtileri ve yutma refleksini engelli.
kas şekil belirtiler o kadar şiddetli değildir ve hastalık kendisini azaltılmış kas tonu ve duruş bozuklukları tezahür edince. Bu tür rahatsızlıklarla, hastalar sakin bir şekilde yaşlı yaşarlar.
Kalıtım
Pompe hastalığı kalıtsal olarak kabul edilir ve otozomal iletilir - resesif tip. Anne-babası mutasyonel bir genin taşıyıcısı olan bir çocuk hastalığa yakalanır. Bu ebeveynler Pompe sendromlu bir çocuğa sahip olabilir 
olasılık,% 25'tir. Bununla birlikte, sağlıklı çocuk doğurma şansı var.
Bilim zaten insan vücudunda oluyor sendromu olan ancak henüz başarılı olamadı nedenini bulmak bilir.
Daha yüksek riskler, benzer kan akrabaları arasında hastalık ve yakın akrabalık olan insanlar var riskli kişilerde, Öncekilerden takip eder. Modern sınıflandırma
Pompe hastalığında geleneksel ana semptomlar anlaşıldığı yaşa bağlı olarak 4 gruba ayrılmıştır:
- Erken çocukluk dönemi .Karaciğer ve kalbi etkileyen, hayatın ilk aylarından itibaren ortaya çıkan, hastalığın en şiddetli formu. Kural olarak, çocuklar kalp ya da solunum yetmezliği nedeniyle ölen bir yıla kadar yaşamaktadır.

- Geç yavru tip .Yaşamın ilk 3 yılında kendini gösterir ve çok hızlı gelişmez. Hastalar ergenliğe daha yakın kalıyor ve sebep genellikle kalp yetmezliği.
- Çocuk tipi .İlk belirtiler 6 ila 10 yıl arasında ortaya çıkıyor ve ölümcül sonuç 20 yıla yaklaşıyor.
- Yetişkin tipi hastalık .Kendisini 20 ila 40 yıl arasında tezahür eder, yavaş yavaş gelişir ve hasta çok yaşlı yaşama şansı yakalar.
Hastalığı daha ayrıntılı olarak incelemek oldukça zor çünkü nispeten nadir olarak kabul edilir.
Yaşa bağlı olarak semptomatoloji ve klinik
Farklı yaşlarda hastalık çeşitli belirtilerle kendini gösterir. En şiddetli ve kısa süren hastalık, erken infantil türüdür.
Yaşamın ilk aylarında hastalığın semptomları:
- kas zayıflığı;
- düşük motor aktivitesi;
- gecikmiş fiziksel gelişim;
- açık bir sebep olmaksızın heyecanlanma ve sıkça ağlama;
- solunum sistemindeki bozukluklar;
- karaciğer büyümesi ve kalp kasının proliferasyonunu;
- , yutkunma ve emme reflekslerini ihlal etmek, dilinde bir artış.

Geç bebeklik ve juvenil formun semptomları( 3 ila 10 yıl arası):
- , alt ekstremitelerdeki kas tonusunu kademeli olarak diğer kaslara çevirerek azalttı;
- hareketlerin koordinasyonunda bozulmuş;
- solunum sisteminde sorunlar;
- ağırlık eksikliği;
- kalp, dalak ve karaciğer büyüklüğünde artış.
Hastalığın erişkin veya geç bir formunun semptomları( 20 yaşından itibaren):
- uykusuzluk veya artan uyuşukluk;
- sık sık baş ağrıları;
- kas güçsüzlüğü ve şiddetli nefes darlığı;
- skolyoz.
Hastalığın bu formu en az tehlikeli olarak düşünülür ve ölüm oranı çocukluk çağında hastalığın gelişmesinden çok daha düşüktür.

Teşhis arama aşamaları
 Hastalığın tespiti birkaç aşamada gerçekleştirilir.
Hastalığın tespiti birkaç aşamada gerçekleştirilir.
Önce, başlıca tanı yöntemleri izlenir ve hastalığın türüne ve evresine göre ek çalışmalar atanabilir.
Tip 2 glikojenozu saptamanın ana yöntemleri şunlardır:
- anamnezi;
- kan serumunun laboratuvar testleri;
- klinik semptomların değerlendirilmesi;
- Alt ekstremite kaslarının ve vücudun diğer bölümlerinin MR görüntüsü;
- EKG ve EchoCG göğüs röntgeni.
Ana teşhis aktivitelerini yaptıktan sonra, 
- genetik defektler için DNA araştırmaya ayrılmıştır;
- biyopsi ve kas dokusunun mikroskobik analizi;Alfa-glukosidaz aktivitesinin seviyesini belirlemek için
- kan testi.
Hastalığın geç formuna gelince, o zaman bu tür yöntemleri uygulayın:
- polisomnografi;
- akciğer kapasitesinin belirlenmesi;
- iskelet kası elektromiyografi.
Yapılan çalışmaların sonuçlarına ve hastanın genel durumunun değerlendirilmesine dayanarak tedavi önerilir.
Teşhis
yapılırken yapılması gerekenler Öyleyse, hayal kırıklığı yaratan bir teşhis koyulur ve kuşkusuz olur. Sırada ne var? Bu durumda, aşağıdaki eylem algoritmalarını takip etmek gerekir:
- aşağıdaki dokümanlardan bir paket hazırlar: Teşhis, önerilen ilaçlar ve bunların tanıtılma şekli hakkında bilgi içeren tıbbi kartın özü;tıp komisyonlarının protokolleri;engellilik için sevk;
- , tedavi başvurusunun hazırlanması için belirtilen listeyi doktorunuza devretmek;
- , gerekli ilaçların alınması için bir başvuruyla sağlık makamlarını ziyaret;
- yardım için bir kamu kuruluşuna bir başvuru yazın;
- başvurunun reddedilmesi halinde, Sağlık Bakanlığına bildirmek ve hukuki yardım almak gerekiyor.
Modern tıbbın
 'yi sunabileceği Nedir Yukarıda belirtildiği gibi, Pompe hastalığının tedavisi mümkün değildir. Bununla birlikte, bilim bu sorunun birkaç yıldır ele alındı ve bazı başarılar yapıldı.Bugün, işlemi yavaşlatabilir ve hastanın durumunu rahatlatabilirsiniz. Modern tıp Rusya'da ne sunuyor?
'yi sunabileceği Nedir Yukarıda belirtildiği gibi, Pompe hastalığının tedavisi mümkün değildir. Bununla birlikte, bilim bu sorunun birkaç yıldır ele alındı ve bazı başarılar yapıldı.Bugün, işlemi yavaşlatabilir ve hastanın durumunu rahatlatabilirsiniz. Modern tıp Rusya'da ne sunuyor?
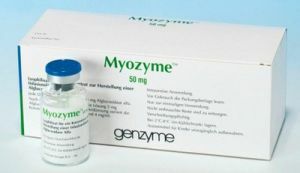
Rusya vatandaşlarına sunulan tek ilaç, intravenöz idare için Mayozaim'dir. Bu, ömür boyu süren bir çare ve eylemi, hastalığın etkilenen organ ve sistemlerin işlevlerini yerine getirmeyi amaçlıyor.
İlaç, her yaşta ve hastalığın her biçiminde kullanılabilir. Aşağıdaki olumlu etkiler kullanım sonucunda kaydedilir:
- , kemiklerin ve kasların durumunu iyileştirir;
- solunum sisteminin çalışmasını stabilize eder;
- motor aktivitesini arttırır.
Mayozeim'in uygulanması ölüm riskini% 99 azaltır ve solunum sisteminin çalışmasını% 92 oranında iyileştirir. Bununla birlikte, bazı hastalarda, ilacın aşağıdaki yan etkileri gözlenebilir:
- kusma ve mide bulantısı;
- deride allerjik bulgular;
- nefes darlığı ve taşikardi.
İlacın dezavantajı, yüksek fiyat olmasıdır. Bununla tedavi yürütme imkanı yoksa, doktorlar hastanın durumunu daha ucuz ve daha az etkili ilaçlarla düzeltmektedirler.
Prognoz ve
önlenmesi Pompe hastalığının prognozu hastalığın şekline bağlıdır. Erken çocukluk döneminde çocuklar bir yaşına gelmeden ölürler.Ölüm nedeni kalp yetmezliği.
Geç bebek ve genç formu hastanın 20 yaşına gelmesine izin verecektir.Ölüm, kardiyopulmoner yetmezlik sonucu oluşur.
Hastalığa 20 yılda tanı konanlar için en olumlu prognoz. Bu insanlar yaşlılıkta yaşayabilir, ancak hemen hemen herkes bir sakatlığa sahiptir.
Önleme konusunda mevcut değil. Bu genetik bir hastalıktır ve sağlıklı bir çocuğun benzer bir hastalık vakası olan bir ailede doğduğuna dair bir garanti yoktur. Hasta bir bebeğin doğum olasılığını yalnızca uygun bir muayene ile belirleyebilirsiniz.