angelmanův syndrom( „happy loutkové“ syndrom „Petržel Syndrome“) - je vzácné neurodevelopmental porucha související s chromozomální abnormality.
angelmanův syndrom( „happy loutkové“ syndrom „Petržel Syndrome“) - je vzácné neurodevelopmental porucha související s chromozomální abnormality.
Přibližně tisíc novorozenců má jedno dítě s touto patologií.U pacientů s fyzickou a duševní opoždění vývoje, poruchy spánku, záchvaty, křeče, trhavé pohyby, soukromá bezdůvodné smích.
Děti s touto patologií vždy vypadají velmi šťastně.
Content
- historie studium patogeneze
- onemocnění a způsobuje
- genetické mutace zvyšují riziko
- skupiny příznaků klinického obrazu
- onemocnění při diagnóze detail
- a včasná identifikace
- foto a video materiálu na předmět
- Zlepšení stavu a života pacientů
- funkcí vzdělávání a přizpůsobení
historiestudující nemoci
nemoc je pojmenována po Harrym Angelmanovým, britský pediatr, který nejprve diferencované syndrom v roce 1965. poté,a byl jmenován syndrom šťastné loutkové, ale dnes tento termín nepoužívá, protože to bylo považováno za hanlivý.
Dr. Angelman se podílel na léčbě několika dětí s podobnými příznaky a navrhl obecnou diagnózu. Prokázání diagnózy a získání přesných dat v té době nebylo možné, kvůli nedostatku technologií, které jsou dnes k dispozici. Jeho teorie doktor odráží v článku nazvaném "Dětské loutky".
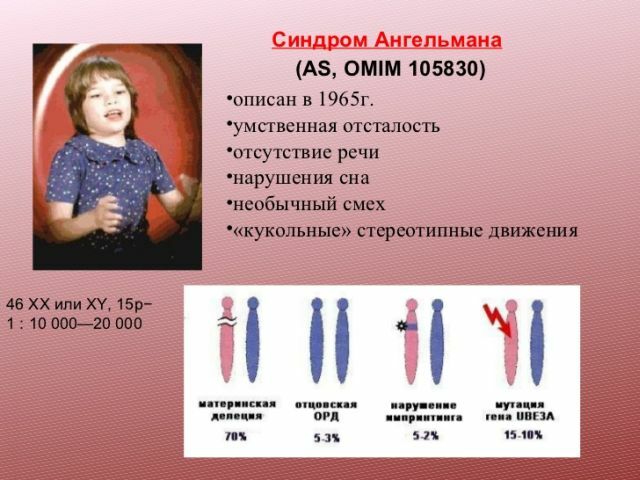
V té době publikace nezpůsobila velký veřejný zájem a byla brzy zapomenuta. Opět se připomínalo v osmdesátých letech, kdy se objevily nezbytné metody výzkumu. Vědci zjistili, že většina dětí se syndromem nemá malou část 15 chromozomů.
Patogeneze a způsobuje
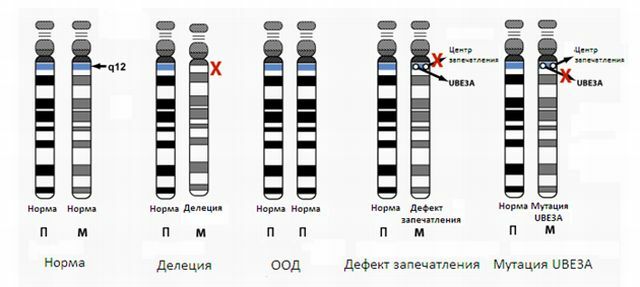
Tato vada vzniká v důsledku delece( výpadek fáze genetického materiálu) úseku chromozomu 15.Mezi další důvody, často nazývaných odnoottsovskuyu Dis( otcovské dědičnosti dvou kopií chromozomu namísto kopií z obou rodičů), translokace, nebo mutace jednoho genu.
syndrom může také nastat jako výsledek genové mutace se podílejí na metabolismu ubikvitinu. Pacienti s touto patologií v rodinné anamnéze
Nejvíce je žádné genetické abnormality. U určitého procenta pacientů je však tento syndrom zděděn. Rizika jsou vyšší, pokud rodiče mají chromozomální abnormality. Zajímavý fakt - v případě, že rodina měla dítě s tímto syndromem, pak pravděpodobnost narození dalšího dítěte se stejným patologie, je 1%.
vzhled Angelmanovým syndromem zcela spontánně, a téměř každá rodina může mít dítě s touto nemocí.Neexistují žádné specifické statistické údaje o počtu dětí s tímto syndromem.
Podle různých odhadů se počet dětí se syndromem panenek se pohybují 1 až 10.000 na jeden z dvaceti tisíc. Vědci naznačují, že toto číslo je ve skutečnosti mnohem větší.
genetické mutace zvyšuje riziko vzniku nemocí
Angelmanův syndrom je spojen s přítomností rodičů o dítě různých chromozomálních abnormalit. Mezi takové odchylky se obvykle říká:
- trizomie chromozomů - přítomnost jednoho nebo více dalších chromozomů v sadě chromosomu;
- inverze - obrácení jednoho z chromozomů částí o 180 stupňů, je část chromozomu je vynechán, a geny jsou uspořádány v opačném pořadí;
- mikrodelece , který je výsledkem přizpůsobení a Y chromozomu segmentů mezi výměnou chromozomy, existuje malý počet chromozomů, a tam může být jeden z genů;
- výmaz - nedostatek jedné z částí chromozomů;
- translokace - převod nebo spojovací oblast z jednoho chromozomu do jiného chromosomu;
- duplikace - kopírování části chromozomů, což vede k přebytku genetického materiálu;
- prstencový chromozóm - na chromozomu končí v režimu offline genetický materiál, nově vytvořené konce jsou spojeny v kruhu.mutace
gen, který může způsobit syndrom projeví příznaky
Groups Obvykle je diagnóza angelmanův syndrom je kladen ve věku 3 a 7 let, kdy symptomy se stávají výraznější mezi nimi. Novorozenec syndrom neliší od zdravých dětí, ale během prvních měsíců života, tam jsou problémy s krmením, děti pomalu přibývají na váze, mají problémy se spánkem, zpoždění ve vývoji motoru.
Všechny příznaky petrželového syndromu jsou rozděleny do několika typů.Mají vztah neuralgické, psychické a fyzické kondici pacienta:
- fyzické příznaky zahrnují problémy se zrakem( strabismu, oční atrofie), vyčnívající jazyk, ústa,
 velká vzdálenost mezi zuby, velikost hlavy úměrně menší ve srovnání s ostatními částmi velikosti těla, albinismus a hypopigmentace, nesgibanie klouby při chůzi nohy( tím vznikalo ve srovnání s loutkami), skoliózy.
velká vzdálenost mezi zuby, velikost hlavy úměrně menší ve srovnání s ostatními částmi velikosti těla, albinismus a hypopigmentace, nesgibanie klouby při chůzi nohy( tím vznikalo ve srovnání s loutkami), skoliózy. - pozorovat třes končetin, různé poruchy spánku, hysterických útoků, jehož intenzita je vysoký ve věku tří let, a postupně klesá, jak stárnou, problémy v procesu komunikace( řeč vývoj z prodlení), hyperaktivitou a poruchou pozornosti Mezi neurologickými příznaky.příznaky
- Psychologické jsou obvykle vyjádřeny v přehnaně emocionální chování( konstantní šťastný vzhled, přátelskost), mentální retardace, těžká nemoc.
Detailní klinický obraz
Různí pacienti mohou vykazovat různé příznaky. Tento rozdíl závisí na stupni a typu chromozomální abnormality.příznaky
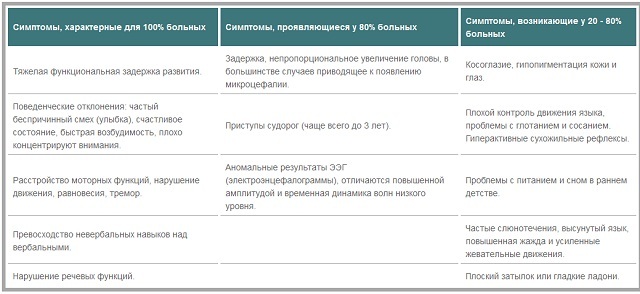
- , které se vyskytují ve všech pacientů :
mohou příznaky s frekvencí jejich výskytu u dětí s Angelmanovým syndromem rozdělit. Patří mezi ně těžkou funkční vývojové zpoždění, problémy s chováním( směje se a usměvavý, bezdůvodně stav štěstí, podrážděnost, snížená koncentrace pozornosti).Také naprosto všichni pacienti mají poruchy v motorických funkcí, poruchy rovnováhy, třes, převaha neverbálních dovedností verbálních, poruchy řeči.
- Symptomy typické pro 80% pacientů s .Zpomaluje růst, což vede k disproporci hlavy vzhledem k ostatním částem těla.Často to vede k rozvoji mikrocefalie. U dětí ve věku do tří let se objevují epileptické záchvaty, pak se stávají méně časté nebo úplně zmizí.Výsledky elektroencefalogramu jsou často anomální, nízkoúrovňové vlny mají zvýšenou amplitudu a časovou dynamiku.
- Příznaky, které se vyskytují u méně než 80% pacientů s .Tyto projevy patří strabismus, sníženou schopnost kontrolovat pohyby jazyka, potíže s polykáním, hypopigmentace kůže a očí, albinismus, mnoho pacientů dochází ke zvýšenému aktivity šlachových reflexů.V raném dětství mají mnozí problémy s výživou a spánkem. Mezi projevy lze také nazývat slinění, vytrháním z jazyka, stálou žízeň.Externí funkce zahrnují přítomnost plochého zátylku a hladkých dlaní.
syndrom projevy mohou být různé.Poruchy spánku, hyperaktivita, konvulzivní syndrom jsou méně časté.Dospělí, kteří mají Angelmanův syndrom, vypadají mladší než jejich vrstevníci.
puberta nastane o něco později, než u osob bez tohoto onemocnění.Pacienti mohou mít vlastní děti, ale s vysokým rizikem pro přenos choroby na potomky.
Mnoho dospělých pacientů trpí nekontrolovaným močením.Často se tvrdí, že existují problémy s motorikou, což má za následek nutnost nosit šaty bez zipů, knoflíků.Mezi dospělými pacienty mají problém s nadváhou, a proto je nutné, aby sledoval dodržování speciální diety.
Děti jsou vnímaví k mluvenému slovu, a porozumět obsahu téměř všech rozhovorech, ale jen zřídka setkat.Často odmítají účastnit rozhovoru, a používat ve svém projevu několik desítek slov.
diagnóza a včasná identifikace
syndrom Doll možné diagnostikovat před narozením, a to prostřednictvím genetického výzkumu patnáctého chromozomu.
Diagnóza může být invazivní a neinvazivní.Invazivní studie v medicíně je dlouhá doba, ale je poněkud riskantní, protože je třeba se proniknout do dělohy a plodové příjmem tekutin.
neinvazivní způsob zahrnuje analýzu krvi matky, ve kterém se dítě provádí analýzu DNA.Na základě této studie, závěry o přítomnosti nebo nepřítomnosti určitých odchylek.
Diagnostics také drží novorozence, který pozorované porušení svalového tonu, opožděným vývojem řeči a motorických dovedností.
je nutné sledovat dětskou tvář, jeho chování, je projevem emocí, pohybů.V případě, že dítě má potíže s ohýbáním končetiny, je třes, chaotické a prudké pohyby končetin, měli byste požádat o radu a pomoc odborníků.
Často diagnóza angelmanův syndrom je kladen ve věku od 3 do 7 let, mezi okamžikem, kdy si všimnete živý projev symptomů nemoci. V závislosti na stupni poškození chromozomu 15, projevy mohou mít různý stupeň závažnosti: někteří pacienti mají potíže i v řeči, zatímco jiní mohou vést nezávislý život.
foto a video materiály na toto téma
Video, stejně jako galerie s fotografiemi dětí a dospělých s diagnózou Angelmanovým syndromem:





zlepšení stavu a života pacientů
Angelmanův syndrom je genetická choroba, zdalekaden není žádné metody obnovení chromozomálních abnormalit, a léčba není možné.Nicméně, existuje mnoho způsobů, jak snížit příznaky, což usnadňuje uvést tyto lidi.
V každém případě je rehabilitační program je individuálně navržen tak, respektive, a příznaky konkrétního pacienta. Odborníci identifikovat čtyři hlavní oblasti léčby:
- recepce antiepileptika a antikonvulziva .Tyto léky pomáhají kontrolovat a snížit četnost útoků
 způsobil onemocnění.
způsobil onemocnění. - Terapeutická cvičení - pomáhá rozvíjet jemnou motoriku a řešení dalších problémů, pohybový aparát. Děti s abnormálními chromozomy rozvíjet pomaleji než jejich zdraví vrstevníci, a to vyžaduje trpělivost ze strany rodiny.
- znakový jazyk .Pacienti se syndromem loutkové málo mluví, ale celkem úspěšně používají znakovou řeč.Školení by mělo začít od velmi raného věku.
- Behaviorální terapie .Tento program nám umožňuje poskytnout správné a efektivní vzdělávání dětí, které mají abnormality, které pomáhají s hyperaktivitou a poruchou pozornosti.
Mnoho lékařů zaznamenává podobnost mezi dětmi s autismem a Angelmanovým syndromem. Američtí vědci pokročili v léčbě, konkrétně při použití intravenózních injekcí s hormonem Secretin. Pozitivní efekt je vyjádřen snížením příznaků nežádoucího chování a zlepšením komunikačních schopností.
Charakteristiky vzdělávání a adaptace
Jak dítě roste touto patologií, symptomy se mění, stávají se méně intenzivní, nebo naopak intenzivnější, staré mohou zmizet a objevit se nové.Vyhlídky ve stavu pacienta velmi závisí na okolnostech, které obklopují pacienta.
Zklidňující prostředí, atmosféra citové náklonnosti poskytuje pacientovi možnost stát se alespoň částečně nezávislým člověkem. S náležitou péčí se známky onemocnění stávají snazšími s časem. Průměrná délka života u lidí s Anghelmanovým syndromem je průměrná.Je třeba si uvědomit pravidelné vyšetření, správnou výživu a dodržování podmínek léčby.
Výuka dítěte ve škole vyžaduje použití inkluzivních programů.Nicméně v Rusku av zemích SNS nejsou tyto programy v současné době zahrnuty do široké praxe. Takové děti potřebují zvláštní přístup, který zahrnuje školení v koncentraci pozornosti. Důležitým požadavkem je také možnost, aby vzdělávací instituce poskytla lékařskou pomoc v případě vhodnosti.
V současné době jsou lidé s chromozomálními abnormalitami obezřetní a opatrní.V posledních letech byly aktivně prováděny různé aktivity zaměřené na šíření poznatků o těchto onemocněních a na vytváření tolerance ve společnosti.