 Pour la première fois, le syndrome de Louis-Bar a été vu et décrit en France en 1941.Depuis lors, sa fréquence d'apparition a sensiblement augmenté et a commencé à se produire dans le monde entier.
Pour la première fois, le syndrome de Louis-Bar a été vu et décrit en France en 1941.Depuis lors, sa fréquence d'apparition a sensiblement augmenté et a commencé à se produire dans le monde entier.
Les statistiques disent, dans la société d'aujourd'hui, 1 personne sur 40 mille personnes a une chance d'avoir ce syndrome.
Son essence réside dans le statut immunitaire innée de l'organisme mal , qui affecte en particulier le T-lien et commence à se manifester dans des changements anormaux dans le corps.
Les personnes atteintes du syndrome sont sujettes à des maladies infectieuses fréquentes et ont également de fortes chances de développer des tumeurs malignes dans tout le corps.
Le plus souvent si le syndrome Louis-Bar commence à se manifester chez les enfants à la naissance, il se heurte à des mortels même sans chance de diagnostiquer ces patients correctement et à temps.
Maladie dans la même proportion affecte les hommes et les femmes, aussi rapidement que possible, détruisant leur système nerveux et la peau.
Causes de
Le syndrome peut survenir au niveau génétique, avec le moindre dysfonctionnement ou anomalie.
Un échec similaire est lourde de dysplasie neuroectodermique, qui est innée chez ces personnes.
La pathologie fait référence aux maladies autosomiques récessives, qui peuvent survenir si les troubles génétiques étaient présents simultanément chez les deux parents.
La maladie tend à changer complètement et à détruire les tissus du cervelet, atteignant même son noyau.
Des situations similaires conduisent à des changements dégénératifs dans le cortex cérébral, ainsi que dans la moelle épinière.
Il est possible de le montrer seulement après un traitement long et difficile des maladies infectieuses, qui ne donnent pas le résultat désiré.
Des troubles immunitaires puissants conduisent à la formation de tumeurs malignes, qui proviennent du système lymphoréticulaire. Les symptômes du syndrome
dans la médecine moderne, la pathologie est rare, mais les médecins craignent la maladie possible le développement de 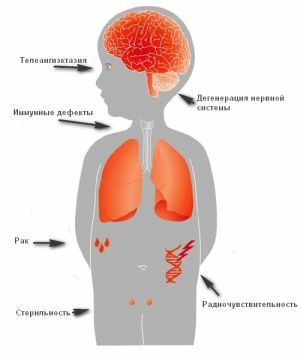 .
.
Puisque cette maladie génétique détruit partiellement ou complètement l'immunité cellulaire, elle a un caractère pathologique et ne peut pas être traitée. Une vie pleine est presque irréelle.
La maladie symptomatique à l'âge adulte ne peut pas être exprimée immédiatement.
Le plus souvent, il est détecté par la détérioration progressive des organes internes, la défaite du système immunitaire, l'absence complète ou partielle du thymus.
Si le syndrome Louis-Bar se développe in utero, affectant le cervelet et l'enfant du cortex cérébral, nouveau-né de la naissance d'un changement dégénérative et condamne à tourmenter le diagnostic .
Si à la naissance, le bébé n'a pas remarqué les premiers signes de la maladie, alors à l'âge de 3-24 mois le syndrome commencera à se manifester assez rapidement.
Le plus souvent, cela s'exprime en l'absence totale de mouvements, une mauvaise coordination, une stagnation du développement mental et des signes extérieurs de développement du visage et des membres.
Il peut être:
- hypotension musculaire;
- strabisme;
- manque de réflexes et de fonctionnalité des muscles et des yeux.
Le syndrome de Louis-Bar se manifeste souvent par des maladies infectieuses persistantes qui touchent les voies respiratoires et les oreilles.
Cela peut être l'otite moyenne, la pharyngite, la bronchite, la sinusite et d'autres maladies.
La pneumonie et la pneumonie ne se manifestent presque jamais. Chaque maladie subséquente a une forme plus aiguë et des complications qui ne se prêtent pas au traitement.
Le signe du symptôme est également considéré comme des astérisques vasculaires, qui peuvent apparaître dans tout le corps à l'âge de 3 ans.
Le plus souvent, cela est associé à l'expansion des capillaires, mais en présence de ce seul symptôme, vous devez rechercher d'autres variantes de maladies possibles.
Quant à l'apparence externe du visage et des yeux, ici commence en premier lieu à manifester des télangiectasies sur le globe oculaire.
est pleine de conjonctivite constante, les symptômes visuels qui peuvent se manifester non seulement devant, mais aussi sur le cou, les joues, les oreilles, les paupières, et même sur les paumes.
En plus de ce code, tout le corps devient sec et squameux, la couverture capillaire est abondante.
Dans les situations les plus négligées, le syndrome peut provoquer des tumeurs malignes, des leucémies et des lymphomes.
Qu'est-ce qui est fait pour le diagnostic?
 Au premier signe ou suspicion d'une maladie de ce type, tout médecin prend rendez-vous et le renvoi à un médecin d'une spécialisation plus étroite.
Au premier signe ou suspicion d'une maladie de ce type, tout médecin prend rendez-vous et le renvoi à un médecin d'une spécialisation plus étroite.
Assez souvent, ces patients sont observés simultanément par plusieurs médecins qui prescrivent un traitement par une consultation conjointe.
Il peut être un immunologiste, dermatologue, ophtalmologiste, neurologue, oncologue et oto-rhino-laryngologiste. Seules leurs consultations conjointes seront en mesure de distinguer ce symptôme d'autres types de maladies rares et dangereuses.
Le diagnostic final dans une telle maladie est toujours posé uniquement par un neurologue s'il dispose de tous les résultats des tests cliniques et des tests de laboratoire.
Le plus souvent, certains indicateurs aident à établir le diagnostic, qui ne correspond pas à la norme. En particulier, le sang peut complètement manquer de lymphocytes, et le taux d'immunoglobuline sera beaucoup plus faible que la normale.
Dans le même temps, absolument aucun anticorps ne sera disponible pour combattre les infections virales et les maladies.
Lorsque le diagnostic final est pour un neurologue sur ses mains, alors vous pouvez prescrire un cours spécifique et un régime de traitement pour un tel patient.
Comment prolonger la vie d'un patient?
Actuellement, malheureusement, le niveau de la médecine n'a pas atteint le niveau de trouver des méthodes efficaces et rapides de lutte contre cette maladie génétique.
Les méthodes de traitement sont encore sujettes à la recherche et à l'étude de nombreux scientifiques. Cependant, pour maintenir le maintien de la vie de tels patients, il est habituel d'utiliser un traitement symptomatique palliatif. 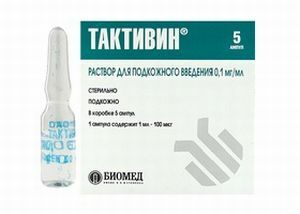
Pour prolonger la vie de ces patients, une immunothérapie spéciale est prescrite, qui peut inclure différentes doses de préparations de T-activatedin et de gamma-globuline.
Dans le même temps, une forte dose constante de préparations de vitamines est obligatoire, qui sont introduites de manière complexe pour maintenir le bon fonctionnement de l'organisme entier.
Si, en même temps, un patient atteint du syndrome de Louis-Bar a une sorte de maladie infectieuse, alors il est traité principalement avec une thérapie intensive pour commencer le processus de maintenir le corps au niveau approprié sans bactéries et virus inutiles.
Selon les troubles observés dans le corps, les médicaments et leurs dosages peuvent varier de manière significative. Souvent, le cours du traitement est complété par des médicaments antifongiques et antiviraux, ainsi que des antibiotiques puissants.
 Inflammation non autorisée et déraisonnable de la psyché sur des maladies inexistantes - névrose hypochondriacale. Que doit-on faire pour traiter le trouble?
Inflammation non autorisée et déraisonnable de la psyché sur des maladies inexistantes - névrose hypochondriacale. Que doit-on faire pour traiter le trouble? L'épilepsie de Jackson diffère des autres types de maladie. Les méthodes de diagnostic et de traitement sont discutées ici.
Prévisions réelles
Parce que le syndrome de Louis-Bar est tout à fait nouveau et complètement inexploré, il est impossible de parler de chances élevées de traitement et encore plus pour le rétablissement du patient.
La pathologie a un pronostic défavorable qui, en fonction de divers facteurs, peut se dérouler au même niveau pendant de nombreuses années et se dégrader rapidement.
Le symptôme le plus commun se trouve dans l'enfance profonde ou à la naissance d'un enfant. L'âge moyen de ces enfants est d'environ 3 ans.
Si les symptômes se manifestent plus tard, ces patients survivent jusqu'à un maximum de 20 ans.
Le plus souvent, la cause de leur mort n'est pas la maladie elle-même, mais la destruction complète de l'immunité et le développement rapide des formations oncologiques dans tout le corps.