Al giorno d'oggi, ci sono un certo numero di orfani( rari) malattie che non sono in grado di gestire la medicina moderna. La possibilità è solo di supportare tali pazienti, in una certa misura migliorare la loro qualità della vita, ma la malattia non ha successo.
Al giorno d'oggi, ci sono un certo numero di orfani( rari) malattie che non sono in grado di gestire la medicina moderna. La possibilità è solo di supportare tali pazienti, in una certa misura migliorare la loro qualità della vita, ma la malattia non ha successo.
malattia di Pompe( glicogenesi generalizzato) attualmente non appartiene alla lista di queste malattie, anche se modi per trattare questa malattia, la scienza non ha ancora trovato. Tuttavia, i pazienti russi hanno la possibilità di essere trattati nel proprio paese e in determinate condizioni, a ricevere assistenza da parte dello Stato, enti pubblici e fondazioni di beneficenza.
Content
- Caratteristiche generali
- Storia e Statistica
- cause e il meccanismo di sviluppo
- Inheritance
- sintomi moderna classificazione
- e la clinica a seconda del
- all'età Stages di ricerca diagnostica
- Cosa fare quando diagnosticato
- Che cosa può offrire moderna medicina previsione e prevenzione
Caratteristiche generali della sindrome di
Pompe colpisce le cellule nervose e muscolari del corpo. Questo accade a causa della mancanza di alfa-glicosidi, che è il risultato di cambiamenti mutazionali a livello genetico.
Se il paziente non riceve un adeguato trattamento, i cambiamenti di struttura tessuto muscolare, dopo di che non è più in grado di funzionare normalmente. Il processo progredisce nel tempo, la lesione si diffonde ad altri gruppi muscolari, che è accompagnata da un forte dolore.
Negli stadi più gravi, i pazienti perdono la capacità di muoversi autonomamente ea spese delle lesioni muscolari nel sistema respiratorio può diventare dipendente da macchine per la ventilazione meccanica.

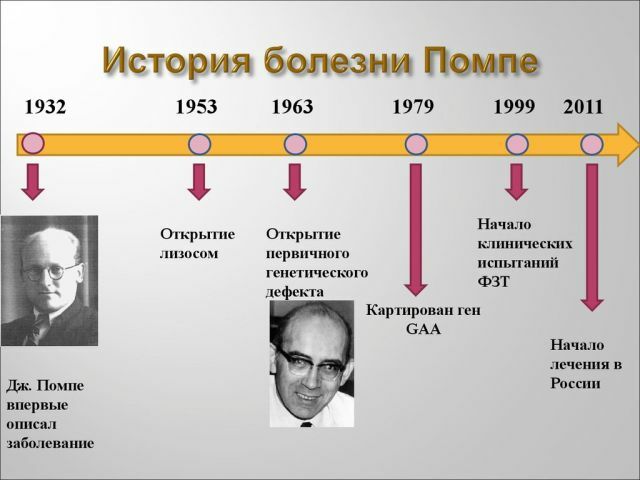
Storia e Statistica
rivelato la malattia era relativamente recente - negli anni '30 del secolo scorso. Per la prima volta è stato descritto da uno scienziato olandese, I. Pompe.malattia
in grado di colpire con uguale probabilità di entrambi i sessi, non è prevalentemente maschile o femminile. La prima forma di malattia si verifica in 1 a 140.000 ai bambini non ancora nati e adulti la malattia di Pompe colpisce 1 su 60.000 persone.
Nel 2006, la metodologia permette di sostenere tali pazienti, e entro il 2013 modi efficaci per trovare e scienziati russi è stato sviluppato negli Stati Uniti. Ma, sfortunatamente, non tutti i pazienti possono permettersi di essere trattati, poiché il costo della terapia è molto alto.
provoca e il meccanismo della malattia sviluppo
sviluppa a causa di glicogeno depositi nel muscolo scheletrico e del miocardio. Questo processo è dovuta a mutazioni nel gene GAA, a causa della quale il muscolo interessato, muscolo cardiaco, del sistema nervoso e del fegato. Il processo va avanti, e si verificano in cellule di vari tipi di danni, tra cui distrofici.
La malattia di Pompe è generalizzata e muscolare. Nel primo caso ci sono cambiamenti in organi quali il fegato ei reni, colpite anche dal sistema muscolare. Inoltre, manifestazioni di insufficienza cardiaca e violazione del riflesso di deglutizione non sono rare.
Quando i sintomi muscolari di forma non sono così gravi, e la malattia si manifesta disturbi del tono muscolare e la postura ridotti. Con questo tipo di disturbo, i pazienti vivono tranquillamente fino alla vecchiaia.malattia
eredità
di Pompe è considerata ereditaria e si trasmette come carattere autosomico - di tipo recessivo. Un bambino i cui genitori sono portatori di un gene mutazionale eredita la malattia. 
La probabilità che tali genitori possano avere un figlio con sindrome di Pompe è del 25%.Tuttavia, ci sono grandi possibilità di dare alla luce bambini sani.
Scienza sa già che cosa sta succedendo nel corpo umano, che soffre della sindrome, ma scoprire la causa non è ancora riuscita.
Da quanto precede, in soggetti a rischio che hanno le persone con malattia simile tra i parenti di sangue, e più la parentela, maggiore è il rischio.moderna classificazione malattia
Pompe convenzionalmente divisi in 4 gruppi a seconda dell'età in cui primario manifesta sintomi:
- Primo tipo infantile .La forma più grave della malattia, che si manifesta fin dai primi mesi di vita, che colpisce il fegato e il cuore. Di regola, i bambini non vivono fino a un anno, morendo di insufficienza cardiaca o respiratoria.

- Tipo infantile tardivo .Si manifesta nei primi 3 anni di vita e si sviluppa non così rapidamente. I pazienti muoiono più vicini all'adolescenza e la causa è più spesso l'insufficienza cardiaca.
- Tipo giovanile .I primi sintomi compaiono da 6 a 10 anni e il risultato letale si avvicina ai 20 anni.
- Tipo di malattia per adulti .Si manifesta da 20 a 40 anni, si sviluppa lentamente e il paziente ha abbastanza possibilità di vivere fino a tarda età.
Studiare la malattia in modo più dettagliato è piuttosto difficile, perché è considerato relativamente raro.
Sintomatologia e clinica a seconda dell'età
A diverse età, la malattia si manifesta con vari sintomi. Il più grave e fugace è il primo tipo di malattia infantile.
Sintomi della malattia nei primi mesi di vita: debolezza muscolare
- ;
- bassa attività motoria;
- ha ritardato lo sviluppo fisico;Eccitabilità
- e pianto frequente senza una ragione apparente;Disturbi
- nel sistema respiratorio;
- allargamento del fegato e proliferazione dei muscoli cardiaci;
- violazione dei riflessi di inghiottire e succhiare, un aumento della lingua.

Sintomi di forma tardiva infantile e giovanile( da 3 a 10 anni):
- ha diminuito il tono muscolare negli arti inferiori con una transizione graduale ad altri muscoli;
- compromissione del coordinamento dei movimenti;Problemi
- da parte dell'apparato respiratorio;Mancanza di peso
- ;Aumento di
- delle dimensioni del cuore, della milza e del fegato.
Sintomi di una forma adulta o tardiva della malattia( a partire da 20 anni): insonnia
- o aumento della sonnolenza;
- mal di testa frequenti;Debolezza muscolare
- e grave mancanza di respiro;Scoliosi
- .
Questa forma della malattia è considerata la meno pericolosa e il tasso di mortalità è molto più basso qui che con lo sviluppo della malattia durante l'infanzia.

Stadio di ricerca diagnostico
 Il rilevamento della malattia viene effettuato in più fasi.
Il rilevamento della malattia viene effettuato in più fasi.
In primo luogo, seguono i principali metodi diagnostici e quindi possono essere assegnati ulteriori studi a seconda del tipo e dello stadio della malattia.
I principali metodi per la rilevazione della glicogenosi di tipo 2 includono: anamnesi
- ;Test di laboratorio
- del siero del sangue;Valutazione
- dei sintomi clinici;
- RM dei muscoli degli arti inferiori e di altre parti del corpo;Radiografia del torace
- ECG ed EchoCG.
Dopo aver svolto le principali attività diagnostiche, l'
- è incaricato di studiare il DNA per difetti genetici;Biopsia
- e analisi microscopica del tessuto muscolare;Analisi del sangue
- per determinare il livello di attività dell'alfa-glucosidasi.
Quando si tratta della forma tardiva della malattia, applicare i seguenti metodi: polisonnografia
- ;Determinazione
- della capacità polmonare;Elettromiografia
- dei muscoli scheletrici.
Sulla base dei risultati degli studi condotti e della valutazione delle condizioni generali del paziente, è prescritto il trattamento.
Cosa fare quando viene fatta la diagnosi
Quindi, viene posta una diagnosi deludente e non c'è dubbio. Cosa fare dopo? In questo caso è necessario seguire il seguente algoritmo di azioni:
- prepara un pacchetto dei seguenti documenti: un estratto della scheda medica, che dovrebbe contenere informazioni sulla diagnosi, i farmaci raccomandati e la modalità della loro introduzione;protocolli di commissioni mediche;referenza per disabilità;
- trasferire l'elenco indicato al medico curante per la preparazione della domanda di trattamento;
- visita le autorità sanitarie con una domanda per l'acquisto di farmaci necessari;
- scrive un'applicazione per un'organizzazione pubblica per assistenza;
- se la domanda è stata respinta, è necessario informare il Ministero della salute e chiedere assistenza legale.
Cosa può offrire la medicina moderna
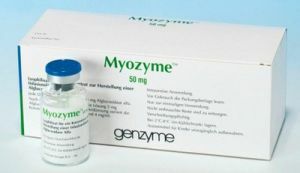 Come accennato in precedenza, è impossibile curare la malattia di Pompe. Tuttavia, la scienza ha affrontato questo problema da diversi anni e alcuni risultati sono stati raggiunti. Oggi puoi rallentare il processo e alleviare le condizioni del paziente. Cosa offre la medicina moderna in Russia?
Come accennato in precedenza, è impossibile curare la malattia di Pompe. Tuttavia, la scienza ha affrontato questo problema da diversi anni e alcuni risultati sono stati raggiunti. Oggi puoi rallentare il processo e alleviare le condizioni del paziente. Cosa offre la medicina moderna in Russia?
L'unico farmaco disponibile per i cittadini russi è Mayozaim per somministrazione endovenosa. Questo è un rimedio per tutta la vita, la sua azione è volta a ripristinare le funzioni di organi e sistemi che sono stati colpiti dalla malattia.
Il farmaco può essere utilizzato a qualsiasi età e in tutte le forme della malattia. I seguenti effetti positivi sono noti come risultato del suo utilizzo:
- migliora le condizioni delle ossa e dei muscoli;
- stabilizza il lavoro del sistema respiratorio;
- aumenta l'attività motoria.
L'applicazione di Mayozeim riduce il rischio di morte del 99% e migliora il lavoro del sistema respiratorio del 92%.Tuttavia, in alcuni pazienti, si possono osservare i seguenti effetti indesiderati del farmaco: vomito e nausea da
- ;
- manifestazioni allergiche sulla pelle;Mancanza di respiro e tachicardia
- .
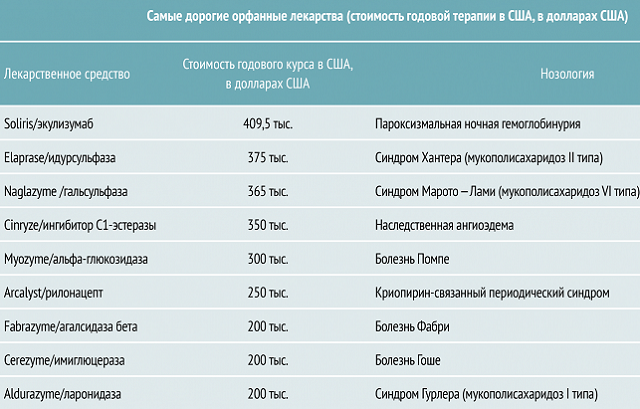
Lo svantaggio del farmaco è il suo alto prezzo. Se non vi è alcuna possibilità di effettuare un trattamento con esso, i medici correggono le condizioni del paziente con l'aiuto di farmaci meno costosi e meno efficaci.
Prognosi e prevenzione dell'
La prognosi della malattia di Pompe dipende dalla forma della malattia. Nel primo tipo infantile, i bambini muoiono prima che raggiungano l'età di un anno. La causa della morte è l'insufficienza cardiaca.
La forma tardiva infantile e giovanile permetterà al paziente di raggiungere l'età di 20 anni. La morte si verifica a seguito di insufficienza cardiopolmonare.
La prognosi più favorevole per coloro con cui la malattia è stata diagnosticata dopo 20 anni. Queste persone possono vivere fino alla vecchiaia, ma quasi tutti hanno una disabilità.
Per quanto riguarda la prevenzione, non esiste. Questa è una malattia genetica e non vi è alcuna garanzia che un bambino sano sia nato in una famiglia in cui vi siano stati casi simili di malattia.È possibile determinare il grado di probabilità della nascita di un bambino malato solo attraverso un esame appropriato.