 Gallervarden-Spatz disease( stiffness progressive ) is a rare hereditary neurodegenerative disease in which basal ganglia are affected and the accumulation of iron in them.
Gallervarden-Spatz disease( stiffness progressive ) is a rare hereditary neurodegenerative disease in which basal ganglia are affected and the accumulation of iron in them.
Accumulation processes occur in a black substance and in a pale balloon. Also in the cerebral cortex changes are observed in nerve cells, the appearance of pigmented neuroglia, and glial cells similar to glia in Alzheimer's disease.
For the first time the disease was described by German researchers Julius Gallervorden and Hugo Spatz, whose names were named.
However, in modern international medical practice, the disease is called neurodegeneration with deposition of iron in the brain( NBIA).Refusal of the original name is due to the fact that Julius Gallervorden conducted research on euthanasia in fascist Germany.
The disease is considered rare, for 1 million people there are up to 3 people with the disease. This neurodegeneration can lead to Parkinsonism, dystonia, dementia, and death.
Features of the genetic malfunction
Gallervorden-Spatz disease is a hereditary genetic pathology. The disease can be both family and sporadic( accidental), but it is always inherited. The disease refers to the autosomal recessive type of inheritance.
For transmission of the disease, both parents must be heterozygous carriers of the disease and have one mutated allele.
The disease is caused by a mutation in the pantothenate kinase gene( PANK2) which is located in the 20 chromosome at the locus 20p12.3-p13.The pantothenate kinase gene is responsible for the coding of the pantothenate kinase 2 protein, which in turn inhibits the accumulation of the amino acid cysteine and panthetein. 
As a result, chemical compounds are formed with iron ions, which have a negative effect on proteins and give rise to the processes of peroxidation, which leads to a programmed cell death of neurons. Instead of dead neurons glial tissue grows( auxiliary cells of the nervous tissue).
Pathological processes occur in the pallid sphere, red nuclei and black matter( nigra substances).They accumulate a substance of a greenish-brown color, which contains iron. In addition, spheroid neuro-axial formations( axonal dilations with proliferation of tubular and membrane structures) are observed in the white matter of the brain, the cerebral cortex, peripheral nerve trunks, and the spinal cord.
Forms of pathology
Three forms are distinguished in the Gallervorden-Spatz syndrome. The division is based on the age at which the development of the disease begins:
- Pediatric form .The onset of the development of the disease occurs at the age of up to 10 years.
- Juvenile( adolescent) form of .The disease develops between 10 and 18 years.
- Adult( atypical) form of .The disease manifests after 18 years.
Features of the clinical picture
Symptoms of this pathology can vary in each case, and the manifestation of symptoms depends on the form of the disease in the individual.
The pediatric form of Gallervorden Spat appears between the ages of 5 and 10 years. It is considered a classic form of the disease.
In the vast majority of cases( 90%), the disease begins with torsional dystonia, which affects the muscles of the legs. The patient has muscle contraction, walking is difficult, gait changes. Then the muscles of the face, pharynx and trunk are affected.
Possible presence of blepharospasm, spasm of hands, spastic torticollis, facial hemispasm. One third of patients have muscle rigidity, hypokinesia, tremor( signs of parkinsonism syndrome).
Typical signs of the infantile form of the disease also include:
- epileptic syndrome;
- aggressiveness;
- mental retardation( due to impaired memory and attention);
- asociality;
- visual disturbances( optic atrophy, retinal degeneration);
- mental disorders.
The adolescent form develops and proceeds more slowly. At the beginning of the disease, 
- focal torsion dystonia( most often affects the muscles of the limbs and maxillofacial muscles);
- neuropsychological disorders;
- behavioral disorders;
- intellectual disorders.
Atypical form of the disease is the most rare( 15% of cases), and it differs from the first two forms. For this form, the most common symptoms are:
- Parkinsonism syndrome with the inability to maintain balance in the standing position;
- involuntary movements in different parts of the body, dystonia, chorea and athetosis( delayed and gusty movements), hemiballism( sweeping motion), myoclonia( not prolonged muscle spasms);
- dementia;
- epileptic syndrome;
- depressiveness, aggression;
- pathological reflexes that arise due to destructive processes in neurons of the brain.
Diagnostic criteria and procedures
The diagnosis of "Gallervorden-Spatz disease" may be problematic for neurologists due to polymorphism of symptoms.
The main criteria for diagnosis:
- onset of illness to 30 years;
- is a typical picture in MRI results;
- permanent development of symptoms;
- extrapyramidal syndromes;
- pyramid signs;
- epileptic seizures;
- cognitive impairment;
- retinal pigmentary degeneration;
- the presence of the disease in the family history.
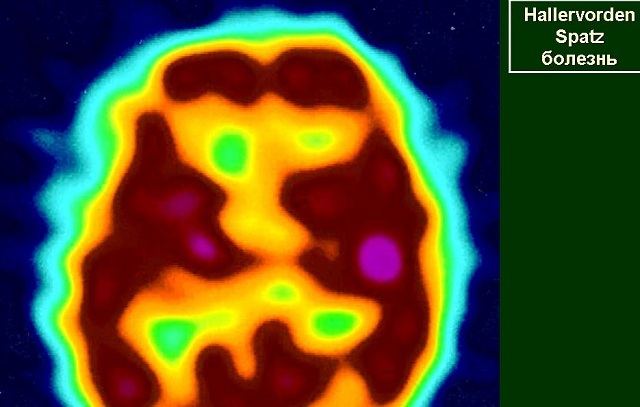
"Tiger eyes" on MRI with contrast is a typical picture of Gallerwarden Spatz disease
The disease must be differentiated from other diseases that have a similar clinical picture: Huntington's chorea, Wilson-Konovalov's disease, choreoacanthocytosis, Machado-Joseph disease.
For diagnosis, the patient is referred for such examinations:
- Study of the neurological status of in a neurologist, neurologist.
- Electroencephalography .
- Consultation of ophthalmologist .It is possible to conduct direct ophthalmoscopy, visual acuity check.
- Genetic studies of , to determine the type of inheritance.
- DNA studies of ( detection of mutations in the PANK2 gene).
- Positron Emission Tomography of the Brain .It is performed to detect a decrease in blood circulation in the fronto-parietal lobes, striatum, pallid sphere.
- Magnetic resonance imaging of the brain .It is required to detect the "tiger's eye" - a hyperintense oval site in the hypo-intensive zone, which arises from the accumulation of iron. There is no unified theory about the development of the "eye of a tiger", there are suggestions of its occurrence before the onset of clinical manifestations of the disease, and its appearance in the years after the onset of the disease. Also MRI allows to detect spheroid neuro-axial formations.
How can modern doctors help?
In modern medicine, there are no treatments that can prevent or stop the Gallervorden-Spatz disease.
Therapy is aimed at alleviating and reducing the intensity of symptoms:
- Parkinsonism syndrome is prescribed dopamine agonists ( Pronoran, Pramipexol, Mirapex, Piribedil), amantadines
 ( Simmetrel, Midantan).However, the resistance of the syndrome to treatment is often observed.
( Simmetrel, Midantan).However, the resistance of the syndrome to treatment is often observed. - valproate ( Convullex, Depakin, Enkorat), benzodiazepines ( Clonazepam, Diazepam) are used to arrest hyperkinesis.
- muscle relaxants ( Midokalm, Baclofen) are used to relieve spasticity of muscles.
- Epileptic seizures are removed with by valproate , Tomapaksom.
- In cases of cognitive impairment, Gliatilin, Neuromidine is used.
- For the treatment of mental disorders, the use of antipsychotics ( clonazepam, quetiapine, raspolent), antidepressants ( dapoxetine, citalopram, venlafaxine) is recommended.
New methods of treating the disease appear. These include therapy by the introduction of pantothenic acid, magnetic stimulation of the brain( pale ball).
Sadly, but not hopelessly
With Gallervorden-Spartz disease, the symptoms are constantly progressing. The most difficult is the child's form of the disease. Full disability of the individual comes in 10-15 years from the moment of the first manifestation of clinical manifestations.
The most favorable development of the disease is predicted in the adult form of the disease. Especially in case of mild dementia.
Therapy allows to preserve the relative quality of life of the patient and his ability to self-service. Life expectancy at an atypical form of Gallervorden-Spatz disease can be more than 20 years.