A síndrome de Angelmann( síndrome de "fantoche feliz", "síndrome da salsa") é uma doença neurogênica bastante rara associada a anormalidades cromossômicas.
Aproximadamente mil recém nascidos têm uma criança com esta patologia. Nos pacientes, há um atraso físico e intelectual no desenvolvimento, distúrbios do sono, convulsões, movimentos violentos e risos privados e sem causa.
Crianças com esta patologia sempre parecem muito felizes.
conteúdo
estudo histórico da doença
patogênese e provoca
mutações genéticas aumentam o risco de grupo
dos sintomas do quadro clínico doença
no diagnóstico detalhe
e identificação atempada
Foto e materiais de vídeo sobre a
assunto melhoria da condição e da vida dos Recursos pacientes
de educação e adaptação
Históriaestudando a doença
A doença tem o nome de Harry Angelman, um pediatra britânico que primeiro diferenciou essa síndrome em 1965. Togde ele recebeu o nome da síndrome de um fantoche feliz, mas hoje este termo não é usado, já que foi considerado desprezível.
O Dr. Angelman esteve envolvido no tratamento de várias crianças com sintomas similares e sugeriu um diagnóstico geral. Para provar o diagnóstico e obter dados precisos naquele momento não era possível, devido à falta de tecnologias disponíveis hoje. Suas teorias que o médico refletiu em um artigo chamado "fantoches para crianças".
Naquela época, a publicação não causou muito interesse público, e logo foi esquecida. Mais uma vez, foi lembrado na década de oitenta, quando os métodos de pesquisa necessários apareceram. Os cientistas descobriram que a maioria das crianças com síndrome não tem uma pequena parte dos 15 cromossomos.
Patogênese e causas de
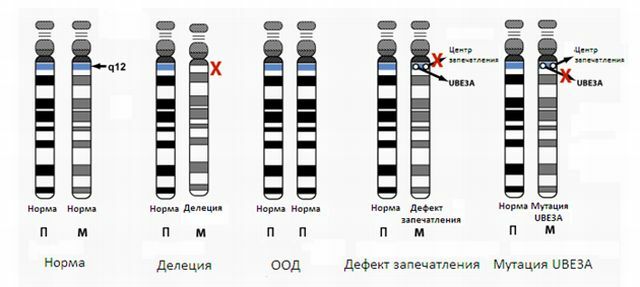
Este defeito decorre da exclusão( perda de material genético) do segmento 15 do cromossomo. Outras causas são muitas vezes chamadas de disomia de pai único( a herança de duas cópias paternas do cromossomo, em vez de cópias de ambos os pais), a translocação ou a mutação de um gene.
Além disso, a síndrome pode aparecer como resultado da mutação do gene envolvido no metabolismo da ubiquitina.
A maioria dos pacientes com esta patologia na história familiar não possui anormalidades genéticas. No entanto, para uma certa porcentagem de pacientes, esta síndrome é hereditária. Os riscos são maiores se os pais tiverem anormalidades cromossômicas. Um fato interessante é que, se uma criança com essa síndrome nascer em uma família, a probabilidade de ter outra criança com a mesma patologia é de 1%.
A aparência da síndrome de Angelman é bastante espontânea e, em praticamente todas as famílias, uma criança com essa doença pode nascer. Não há dados estatísticos específicos sobre o número de crianças com essa síndrome.
De acordo com várias estimativas, o número de recém-nascidos com a síndrome da boneca varia de um para dez mil para um para vinte mil. Os cientistas sugerem que, de fato, esse número é muito maior.
As mutações genéticas aumentam o risco de
A aparência da síndrome de Angelman está associada à presença de várias anormalidades cromossômicas nos pais de uma criança futura. Entre esses desvios são geralmente chamados:
trissomia de cromossomas - a presença de um ou mais cromossomas adicionais no conjunto de cromossomas;
inversão - reversão de um dos cromossomas porções de 180 graus, a porção do cromossoma é omitido, e os genes são dispostos na ordem inversa;microdeleções
, que é o resultado de segmentos de ajustamento e Y-chromosome entre troca cromossomas, há um pequeno número de cromossomas, e pode não haver um dos genes;
eliminação - a falta de uma região do cromossoma;
translocação - a parte de transferência ou junção de um cromossoma de uma outra cromossoma;
duplicação --se parte dos cromossomas, resultante torna-se supérfluo o material genético;
cromossoma anelar - no cromossoma termina material genético desligada, as extremidades recém-formadas são ligadas em um anel. Mutações genéticas
que podem causar a síndrome desenvolvem sintomas
Grupos Normalmente, o diagnóstico de síndrome de Angelman é colocado entre as idades de 3 e 7 anos de idade, quando os sintomas tornam-se pronunciado.síndrome do bebê recém-nascido não diferem de crianças normais, mas durante os primeiros meses de vida, há problemas com a alimentação, as crianças lentamente ganhar peso, têm problemas para dormir, atraso no desenvolvimento motor.
Todos os sintomas síndrome de salsa são divididos em vários tipos. Eles têm uma relação com a condição neuropática, física e mental do paciente:
Os sintomas físicos incluem problemas de visão( estrabismo, atrofia óptica), língua protuberante, de boca larga, grande distância entre os dentes, o tamanho da cabeça proporcionalmente menor em relação a outras partes do tamanho do corpo, albinismo e hipopigmentação, articulações nesgibanie quando andam pernas( daí surgiu em comparação com bonecos), escoliose.
observar tremor nos membros, uma variedade de distúrbios do sono, ataques histéricos, cuja intensidade é alta na idade de três anos, e diminui gradualmente à medida que envelhecem, os problemas no processo de comunicação( atraso no desenvolvimento da fala), hiperatividade e déficit de atenção Entre os sintomas neurológicos .sintomas
psicológicos são geralmente expressos em um comportamento excessivamente emocional( aparência constante feliz, simpatia), retardo mental, doença grave.
quadro clínico em
detalhe pacientes diferentes podem manifestar sintomas diferentes. Esta diferença depende do grau e tipo de anormalidades cromossômicas.
pode dividir os sintomas com a freqüência de seu aparecimento em crianças com síndrome de Angelman: sintomas
que ocorrem em todos os pacientes .Estes incluem grave atraso funcional do desenvolvimento, problemas comportamentais( rindo e sorrindo, sem razão, o estado de felicidade, irritabilidade, diminuição da concentração de atenção).Além disso, absolutamente todos os pacientes têm distúrbios na função motora, perturbação do equilíbrio, tremor, predomínio de habilidades não-verbais de distúrbios verbais, fala. Os sintomas
típicos para pacientes 80%.Nanismo, levando a distorções de cabeça em relação a outras partes do corpo. Isso muitas vezes leva ao desenvolvimento de microcefalia. Crianças menores de três anos de convulsões muitas vezes se manifestam, então eles se tornam menos frequentes ou desaparecer completamente.resultados de EEG frequentemente anómala, baixas ondas aumentaram amplitude e dinâmica temporal.sintomas
decorrentes, pelo menos, 80% dos pacientes .Estas manifestações incluem estrabismo, diminuição da capacidade para controlar os movimentos da língua, dificuldade para engolir, hipopigmentação da pele e dos olhos, albinismo, muitos pacientes há aumento da atividade dos reflexos dos tendões. Na infância, muitos têm problemas com alimentação e sono. Entre os monitores também podem ser chamados babando, salientes língua, sede constante. Os recursos externos incluem a disponibilidade de um apartamento de volta da cabeça e as palmas das mãos suaves.
À medida que cresce, as manifestações da síndrome mudam. Doenças do sono, hiperatividade, síndrome convulsiva são menos comuns. Os adultos que têm a síndrome de Angelmann parecem mais jovens do que seus pares.
A puberdade ocorre um pouco mais tarde do que em indivíduos que não possuem essa patologia. Os pacientes podem ter seus próprios filhos, mas existe um alto risco de transmissão da doença para a prole.
Muitos pacientes adultos sofrem de micção descontrolada. Muitas vezes, há dificuldades com as habilidades motoras, o que leva à necessidade de usar roupas sem relâmpagos, botões. Entre os adultos, há um problema de excesso de peso, então você precisa monitorar a adesão a uma dieta especial.
As crianças bem percebem a fala oral e compreendem o conteúdo de quase todas as conversas, mas raramente respondem. Muitas vezes eles se recusam a participar da conversa e usam algumas dúzias de palavras no discurso.diagnóstico
e identificação atempada
Síndrome boneca possível diagnosticar antes do nascimento, por meio de genética do cromossoma XV de pesquisa.
O diagnóstico pode ser invasivo e não invasivo.estudos invasivos em medicina há um longo tempo, mas é um pouco arriscado, pois você precisa penetrar no útero e ingestão de líquido amniótico.
Um método não invasivo envolve um exame de sangue da mãe, no qual o DNA do bebê é analisado. Com base neste estudo, são tiradas conclusões sobre a presença ou ausência de certas anormalidades.
O diagnóstico também é realizado em recém-nascidos com distúrbios do tônus muscular, atraso no desenvolvimento da fala e das habilidades motoras.
É necessário seguir a expressão do rosto da criança, seu comportamento, a manifestação de emoções, movimentos. Se a criança tiver dificuldades em flexionar os membros, há movimentos tremores, caóticos e afiados dos membros, então vale a pena procurar o conselho e assistência de especialistas.
Muitas vezes, o diagnóstico da síndrome de Anghelman é colocado na idade de 3 a 7 anos, quando você pode notar uma clara manifestação dos sinais dessa doença. Dependendo do grau de dano aos 15 cromossomos, as manifestações podem ter uma severidade diferente: alguns pacientes têm dificuldade mesmo na fala, enquanto outros podem levar uma vida independente.materiais de vídeo sobre o assunto
vídeo, bem como uma galeria com fotos de crianças e adultos diagnosticados com síndrome de Angelman
foto e: bonecas síndrome
criança
adultos com
síndrome de Angelman melhoria da condição e da vida dos pacientes
síndrome de Angelman é uma doença genética, de longedia não há métodos para restaurar anormalidades cromossômicas, e o tratamento é impossível. No entanto, há um grande número de maneiras de reduzir os sintomas, o que facilita a condição dessas pessoas.
Em cada caso, o programa de reabilitação é desenvolvido individualmente, de acordo com os sintomas e condições do paciente individual. Especialistas identificam quatro áreas principais de terapia:
Admissão antiepilépticos e anticonvulsivantes .Esses medicamentos ajudam a controlar e reduzir a freqüência de convulsões causadas pela doença por .
Treino físico terapêutico - ajuda a desenvolver habilidades motoras finas e a resolver outros problemas do aparelho motorizado. Crianças com anormalidades cromossômicas desenvolvem-se mais devagar do que seus pares saudáveis, e isso exige paciência da parte da família.
Sign Language .Os pacientes com a síndrome de marionetes falam pouco, mas com bastante sucesso usam a linguagem gestual. O treinamento deve começar desde uma idade muito jovem.
Terapia comportamental .Este programa permite que você dê uma educação correta e efetiva para crianças com anormalidades, ajudará a lidar com hiperatividade e déficit de atenção.
Muitos médicos observam a semelhança entre crianças com autismo e síndrome de Angelman. Cientistas americanos fizeram progressos no tratamento, nomeadamente o uso de injeções intravenosas com hormônio secretin. O efeito positivo é expresso na redução dos sinais de comportamento indesejável, bem como na melhoria das habilidades comunicativas.
Características da educação e adaptação
À medida que a criança cresce com esta patologia, os sintomas mudam, tornam-se menos intensos ou, ao contrário, mais intensos, os velhos podem desaparecer e novos podem aparecer. As perspectivas na condição do paciente dependem em grande parte das condições que envolvem o paciente.
Um ambiente benevolente, a atmosfera de afeição afetiva proporciona ao paciente a chance de se tornar, pelo menos parcialmente, uma pessoa independente. Com o devido cuidado, os sinais da doença tornam-se mais fáceis com o tempo. A expectativa de vida em pessoas com síndrome de Anghelman é média.É necessário lembrar sobre exames regulares, nutrição adequada, bem como a observância das condições de terapia.
Ensinar uma criança na escola requer o uso de programas inclusivos. No entanto, na Rússia e nos países da CEI hoje, esses programas não estão incluídos em uma ampla prática. Essas crianças precisam de uma abordagem especial, que inclui treinamento na concentração da atenção. Um requisito importante é também a possibilidade de a instituição educacional prestar assistência médica em caso de ajuste.
Em nossa sociedade no momento, pessoas com anormalidades cromossômicas são cautelosas e cautelosas. Nos últimos anos, várias atividades foram ativamente desenvolvidas com o objetivo de disseminar conhecimento sobre tais doenças, bem como criar tolerância na sociedade.

 A síndrome de Angelmann( síndrome de "fantoche feliz", "síndrome da salsa") é uma doença neurogênica bastante rara associada a anormalidades cromossômicas.
A síndrome de Angelmann( síndrome de "fantoche feliz", "síndrome da salsa") é uma doença neurogênica bastante rara associada a anormalidades cromossômicas. 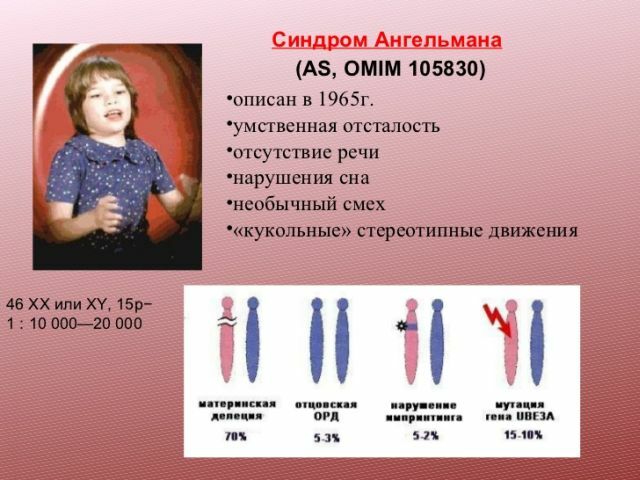
 grande distância entre os dentes, o tamanho da cabeça proporcionalmente menor em relação a outras partes do tamanho do corpo, albinismo e hipopigmentação, articulações nesgibanie quando andam pernas( daí surgiu em comparação com bonecos), escoliose.
grande distância entre os dentes, o tamanho da cabeça proporcionalmente menor em relação a outras partes do tamanho do corpo, albinismo e hipopigmentação, articulações nesgibanie quando andam pernas( daí surgiu em comparação com bonecos), escoliose. 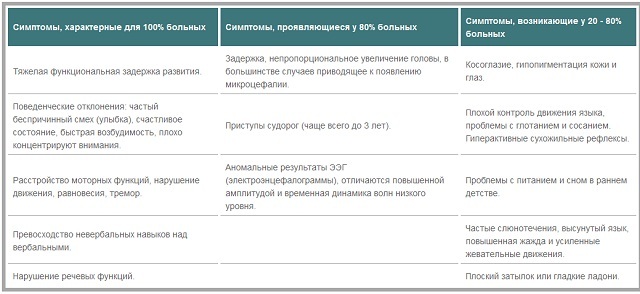
 criança
criança 


 adultos com
adultos com  .
.