Arnold-Chiari Sendromu - doğuştan veya (daha nadiren) edinilmiş omurilik malformasyonukafatasının tabanındaki kemik yapıların yanı sıra.
Son yıllarda, genel morbidite yapısında beyin anomalilerinin %30'a ulaştığı konjenital malformasyonların büyümesi için bir eğilim olmuştur. Neredeyse her zaman, bu bozukluk ikincil serebral değişikliklerle (düşüklük, ventriküler hasar ve diğerleri) birleştirilir.
İçeriği kaydet:
- 1 Arnold Chiari Sendromu nedir?
- 2 epidemiyoloji
- 3 Arnold-Chiari anomalisinin türleri ve dereceleri
-
4 Yetişkinlerde, çocuklarda nedenler ve risk faktörleri
- 4.1 doğuştan
- 4.2 Edinilmiş karakter
- 5 Belirtiler
- 6 İlgili bozukluklar
- 7 komplikasyonlar
- 8 teşhis
- 9 Rahim içi muayene
-
10 Arnold-Chiari malformasyonlu yetişkinlerin ve çocukların tedavisi
- 10.1 konservatif terapi
- 10.2 Cerrahi müdahale
- 11 Sonuçları nelerdir?
- 12 Arnold-Chiari Sendromu Videoları
Arnold Chiari Sendromu nedir?
Arnold-Chiari Sendromu, gövde ve serebellar bademciklerin prolapsusunun ortaya çıktığı beynin ciddi bir patolojisidir. Bunun sonucunda sabit kafa içi basınç sağlayan beyin omurilik sıvısının dolaşımı ve su-elektrolit dengesi bozulur.
Bu, hidrosefali, kafa içi hipertansiyonun kademeli gelişimine ve omurilikte kistlerin ortaya çıkmasına katkıda bulunur (hastaların% 80'ine kadar).
Çoğu zaman, bu ihlal fetal gelişimin doğum öncesi döneminde oluşur. Solunumu ve kan dolaşımını düzenleyen medulla oblongata ciddi şekilde deforme olur ve bundan sorumlu olan beyincik koordinasyon ve kas tonusu, gelişiminde önemli ölçüde geride kalıyor ve yavaş yavaş dikdörtgen üzerinde katmanlaşıyor beyin.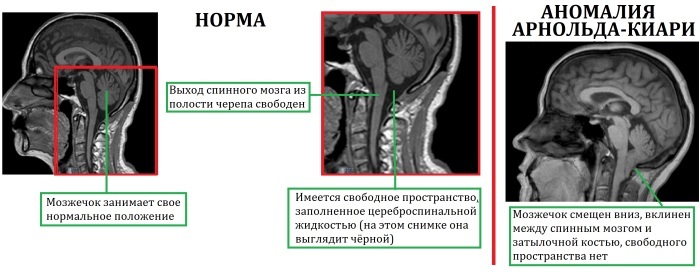
Bu yapıların sıkışması sıklıkla motor ve görme bozukluklarına, epileptik nöbetlere yol açar.
epidemiyoloji
Bu bozukluğun prevalansı oldukça yüksektir - 4-6 bin çocuk başına 1 yenidoğan. Erişkin nüfusta ise 100 binde 3-8 vaka var. nüfus. İlk kez, bu anomali tıpta 19. yüzyılın sonunda ölü yenidoğanların otopsisinden sonra tanımlandı.
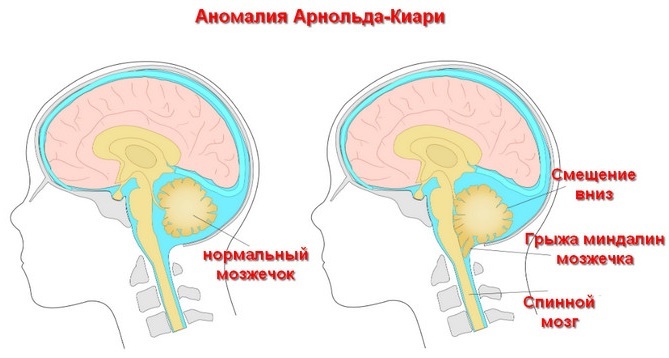
Hastaların %95'inde Arnold-Chiari malformasyonu olduğundan (lat. malus - "kötü" ve formatio - "oluşum") ayrıca bir spinal fıtık oluşumu eşlik eder, daha sonra bu kusur genellikle yanlış tanımlanır. Bu nedenle gerçek prevalans yüksek değerlere ulaşabilir. Vakaların% 14-30'unda hastalık, başka bir nedenle tıbbi muayene sırasında tanısal bir bulgudur.
Arnold-Chiari anomalisinin türleri ve dereceleri
Arnold Chiari Sendromu 4 tipte sınıflandırılan bir hastalıktır:
- Hastalığın seyrinin kolay bir çeşidi. Serebellar bademciklerin, kraniyal boşluğun spinal kanal ile iletişim kurduğu foramen magnum seviyesinin altındaki servikal omurgaya inişi. Çıkıntı 12 mm'yi geçmez. Hastaların %20 kadarında hidrosefali de vardır ve bunların yarısında omurilikte kist gelişir. Bu tür bir bozukluk genellikle ilk kez ergenlik döneminde kendini gösterir, yenidoğanların %50'sinden fazlasında seyri asemptomatiktir. Kadınlarda erkeklerden daha sık teşhis edilir.
- Solucan benzeri bir yapının yerinden çıkması beyincik, medulla oblongata ve IV ventrikülün uzaması. Aynı zamanda hidrosefali ilerler, üçüncü ve dördüncü ventrikülleri birbirine bağlayan Sylvian kanalı daralır, spinal kanalda fıtık oluşur (hastaların% 95-100'ü). Telensefalon (büyük yarım küreler dahil), diensefalon ve orta beyin ve servikal omurganın gelişimindeki bozukluklar da tespit edilir; korpus kallozumun tamamen veya kısmen yokluğu ve lomber omurgadaki omur kemerlerinin aşırı büyümesi. Bu tür bir sapma zaten yenidoğan döneminde teşhis edilir. Doğum öncesi dönemde ve erken yaşta yüksek bir ölüm oranı vardır. Hastalığın ilk 2 tipi en yaygın olanıdır.
-
Toplam taşıma omurgadaki arka beyin yapıları, oksiputta serebral fıtık, belirgin intrakraniyal hipertansiyon, foramen magnumun artan çapı. Hastalar, medullanın bir kısmının kafatasının dışında yer aldığı kafatası ve beyinde büyük bir malformasyona sahiptir. Ayrıca kardiyovasküler patolojiler, anüsün tıkanması ve gastrointestinal sistem ve genitoüriner sistemin diğer anormallikleri nadir değildir. Bu tip sendrom çoğu zaman yaşamla bağdaşmaz.
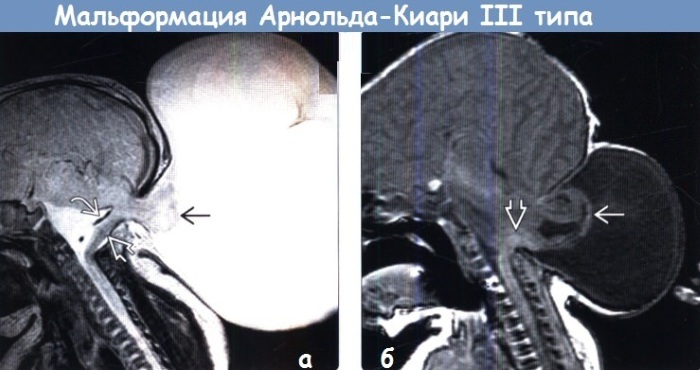
- Azgelişmişlik (hipoplazi) medulla oblongata ile birlikte aşağı doğru yer değiştirmeden beyincik. Bu ihlal yenidoğanın ölümüne yol açar.
Bazı uzmanlar hastalığı 2 gruba ayırır - siringomyeli (omurilikte boşluk oluşumu) ve onsuz. Ayrıca, tip I ve tip II arasında, her iki tipin semptomlarını birleştiren veya hastalığın gelişiminin tamamlanmamış ikinci bir varyantını temsil eden bir geçiş veya sınır evresi vardır.
Yetişkinlerde, çocuklarda nedenler ve risk faktörleri
Bu nörolojik patolojinin gelişim mekanizması, beyinciğin bulunduğu posterior kraniyal fossadaki yapısal değişikliklerin yanı sıra hidrodinamik bozukluklarla ilişkilidir. Bu sendromun nedenleri hala iyi anlaşılmamıştır.
doğuştan
Konjenital defekt, omurilik boşluğunun arka duvarının kafatasının tabanındaki omuriliğin anlage ile kaynaşmasına dayanır. Bu durumda, embriyonik gelişim sürecinde omurilik yukarı çekilmelidir, ancak bu olmaz.
Medulla oblongata ve serebellum, foramen magnumdan spinal kanala doğru "çekilir". Bazen 4. servikal vertebra seviyesinde bulunurlar.
Fetal anormalliklerin gelişimi için risk faktörleri şunlardır:
- hamile bir kadının geç yaşı;
- düşük öyküsü;
- hamilelik sırasında toksikoz;
- plasental yetmezlik, preeklampsi;
- hamile bir kadının zararlı maddelerle teması;
- gebelik sırasında bulaşıcı hastalıklar (cinsel yolla bulaşan hastalıklar dahil).
Bununla birlikte, çalışmalar kadınların neredeyse yarısında hamileliğin komplikasyonsuz ilerlediğini göstermektedir.
Edinilmiş karakter
Yetişkinlerde, omurilikte sık sık delinmeler nedeniyle serebellar yapıların prolapsusu meydana gelebilir. teşhis amaçlı, ayrıca bazı hastalıkların tedavisinde ve omurga olarak gerçekleştirilir. anestezi. Bazı hastalarda bu durum, hidrosefali tedavisi için bir şant sisteminin implantasyonundan sonra gelişir.
Belirtiler
Arnold-Chiari I tipi sendrom uzun süre asemptomatik olabilir.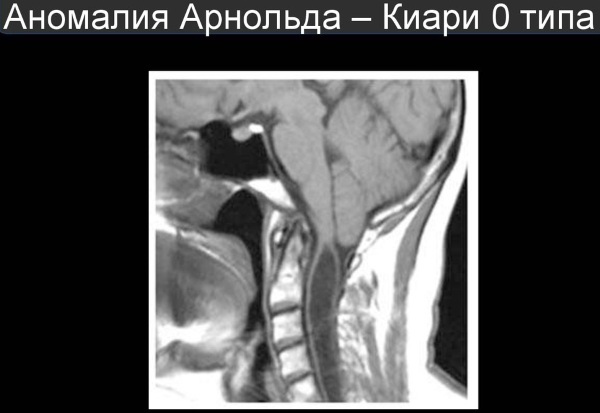
Bu ihlal aşağıdaki belirtilere sahiptir:
- hastalık ergenlerde veya 30-40 yaşlarında kendini gösterir;
- duyarlılık bozuklukları, çoğunlukla ellerde (hastaların %40-76'sı);
- baş ağrısı (hastaların %47-73'ünde gözlenen en yaygın semptom);
- öksürme, hapşırma, karın kaslarının gerginliği ile şiddetlenen boyun ve başın arkasında ağrı;
- sık bayılma, omurilikte bozulmuş kan dolaşımı ile ilişkili düşmeler, bu da kas tonusunda keskin bir azalmaya neden olur; uzuvların uyuşması;
- 10 saniyeden fazla uyku sırasında solunumun sık sık kesilmesi (hastaların %50-70'i), akut solunum yetmezliği;
- uzuvlarda genel halsizlik ve azalmış ton;
- başın değiştirilmiş zorla pozisyonu;
- konuşma ilahisi (ayrı kelimelere bölme);
- salınımlı göz hareketleri, esas olarak aşağı;
- bozulmuş koordinasyon, yürüyüş, fiziksel aktivite; uyumsuzluk, fazlalık veya yetersiz hareket aralığı;
- omurganın eğriliği (hastaların %50-75'i);
- çocuklarda büyüme geriliği ve zihinsel gelişim, bazen erken ergenlik;
- paroksismal öksürük (hastaların %10-14'ü);
- ince motor becerilerin ihlali, uzuvların titremesi;
- işitme bozukluğu (tek taraflı veya iki taraflı);
- konjenital anatomik özellikler - kısa, büyükbaş boyun, huni göğüs, asimetrik kafatasının şekli, boyunda düşük tüylülük sınırı, ayaklarda şekil bozukluğu, meme uçlarında anormallikler, yüksek duruş Omuz bıçakları;
- gözlerde çatallanma, göz bebeklerinin farklı boyutları, görüş alanında kör noktalar, göz kapağının sarkması;
- yutma ihlali, dil kaslarının atrofisi, sık hıçkırıklar;
- nadiren - yavaşlayan kalp hızı, akut kardiyovasküler yetmezlik, hipoglisemi.
Semptomlar lezyonun derecesine bağlı olarak hafif ila şiddetli arasında değişebilir.
Tip II malformasyon ile aşağıdaki belirtiler not edilir:
- yenidoğan döneminde zaten semptomların ortaya çıkması;
- omurga fıtığı;
- hidrosefali (beynin düşmesi, baş çevresinin artması, yenidoğanlarda şişkin fontanel, şaşılık, yüksek uyarılabilirlik, kusma, uyuşukluk, baş ağrısı ve baş dönmesi);
- uyku sırasında da dahil olmak üzere boğulma ve solunum durması atakları, hırıltılı solunum;
- bozulmuş yutma refleksi, yutma güçlüğü (hastaların %70'i);
- kolların ve yüz kaslarının zayıflığı, fiziksel aktivitenin azalması;
- sırtın kavisli konvulsif duruş;
- ses tellerinin parezi sonucu ses kaybı;
- gözbebeklerinin aşağı doğru titremesi.
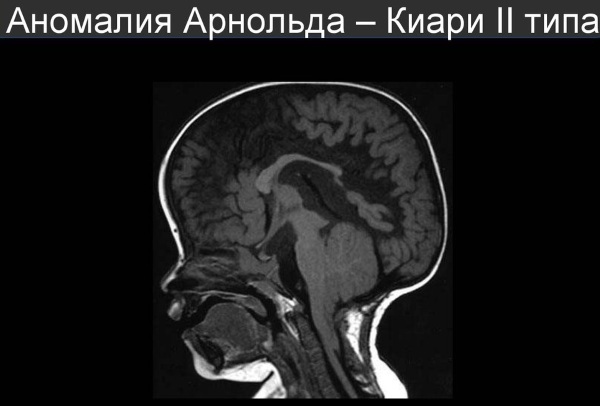
Form III varyantı, aşağıdaki gibi semptomlarla karakterize edilir:
- hidrosefali;
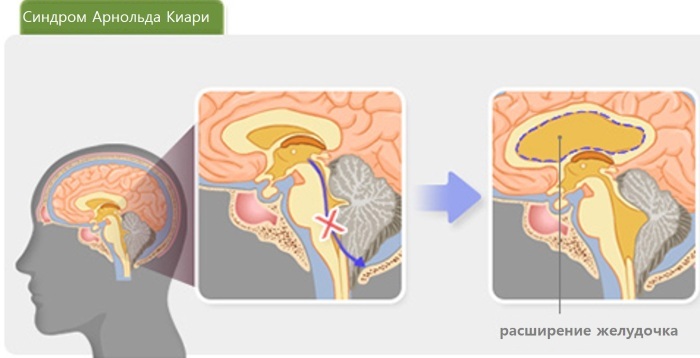
- kardiyovasküler bozukluklar;
- gastrointestinal sistem patolojisi, üriner sistem;
- baş dönmesi;
- hareket ederken istikrarsızlık;
- kulak çınlaması;
- baş ağrısı;
- servikal kaslarda artan ton.
İlgili bozukluklar
Bu sendrom ayrıca genellikle aşağıdaki patolojilerle birleştirilir:
- paranazal sinüslerin kronik iltihabı veya bu bölgede bir kist;
- beyincik ve korpus kallozumdaki tümörler;
- Türk eyer kisti;
- beynin sağ ve sol yarım kürelerini birbirine bağlayan korpus kallozumun tamamen veya kısmen yokluğu;
- aşırı sayıda küçük ve kısmen oluşturulmuş kıvrımlardan oluşan serebral korteksin malformasyonu;
- serebral korteks oluşumunun ihlali (nöronların göçündeki bir değişikliğin neden olduğu çeşitli alanlarda anormal gri madde birikimi);
- kalp ve böbreklerin malformasyonları;
- motor ve otonomik işlevler sağlayan bazal düğümlerin az gelişmişliği;
- çocuklarda kranyal sütürlerin daha erken kapanması, kafatasının hacminin sınırlandırılmasına, deformasyonuna ve intrakraniyal hipertansiyon gelişimine yol açar;
- oksipital kraniyal kemiğin ve birinci servikal vertebranın tam veya kısmi füzyonu (atlas asimilasyonu);
- serebral hemisferlerin subatrofisi (yıkıcı süreçler);
- oksipital kemiğin üst servikal omurganın kafatasının tabanı ile birleşme noktasına doğru yer değiştirmesi (baziler izlenim);
- Maksillofasiyal bölgenin konjenital bir malformasyonunun tespit edildiği Robin anomalisi (alt çenenin az gelişmişliği, yarık damak, dilin geri çekilmesi);
- Kafatasının konjenital bir anomalisinin el ve ayak parmaklarının füzyonu ile birleştiği Aper sendromu;
- Yüzün yapısının değiştiği Williams sendromu, zeka geriliği ve diğer genetik anormallikler gözlenir.
komplikasyonlar
Arnold-Chiari Sendromu erken yaşta yüksek mortalite ile karakterize bir patolojidir. Klinik belirtilerin şiddeti, sinir dokularının sıkışma derecesi ve beyin omurilik sıvısının dolaşımındaki bozulma ile ilişkilidir.
Sendromun II varyantı olan çocuklarda, beyin yapılarının yer değiştirmesine ek olarak, kraniyal sinirler sıkıştırılır, bu da solunum ve yutma fonksiyonlarının bozulmasına yol açar.
Akciğerlere gıda girme ve çoğu durumda erken ölüm nedeni olan aspirasyon pnömonisi gelişme riski artar. Cerrahi, nörolojik bozuklukların azaltılmasına ve hastaların %94'ünde en az 2 yıl stabil bir durumun korunmasına yardımcı olur.
Tip III ve IV'ün kusurları en olumsuz olanlardır, birçok çocuk ameliyat edilemez ve doğumdan sonraki ilk ayda ölür. Hayatta kalanlarda bacaklarda felç, pelvik organların işlev bozukluğu, idrar ve dışkı tutamama ve çoklu malformasyonlar vardır.
teşhis
Patolojiyi teşhis etmek için aşağıdaki araştırma yöntemleri kullanılır:
- makalede daha önce açıklanan karakteristik anomalilerin ortaya çıktığı standart modlarda beynin manyetik rezonans görüntüleme;
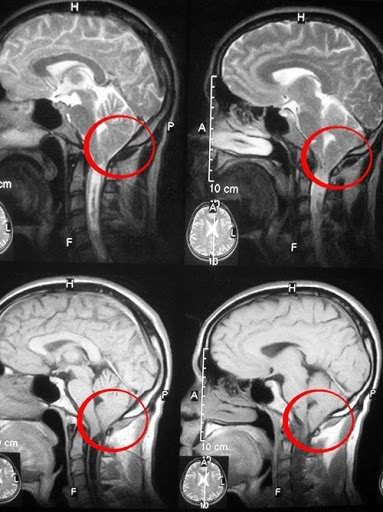
- beyin omurilik sıvısı - beyin omurilik sıvısının hareketindeki ihlalleri belirlemeye izin veren faz kontrast manyetik rezonans görüntüleme (FKMRT);
- nörosonografi (beynin ultrason muayenesi);
- kranyografi (kafatasının kemiklerinin röntgen muayenesi), önceki muayene türü ile birlikte değerlendirmenizi sağlar. posterior kranial fossanın küçülme derecesi, foramen magnumda artış ve diğer intraserebral değişiklikler yapılar.
Rahim içi muayene
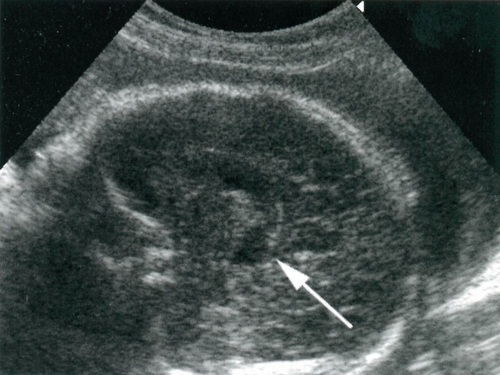
Modern ultrason teşhisi, hamileliğin 17-18 haftasından başlayarak bu patolojiyi ortaya çıkarabilir. Erken aşamalarda, ihlalleri tespit etme doğruluğu %24-45'tir. Çoğu zaman, tanı II-III trimesterlerde yapılır.
Bu süre zarfında hemisferler, beyincik ve onunla ilişkili subaraknoid boşluğun genişleme alanları iyi görüntülendiğinden, taramadan zamanında geçmek önemlidir.
Aynı zamanda, ultrason görüntüleri beyincik şeklindeki ve boyutundaki bir değişikliği, bulanık konturunu, beyincik ve medulla oblongata arasındaki içi boş oluşumlarda bir azalmayı görselleştirir.
Ek teşhis işaretleri de şunlardır:
- hidrosefali;
- beynin ventriküllerinde bir artış ve şekillerinde bir değişiklik (arkaya doğru sivrileşirler);
- cenin başının şekli "limon" ve beyincik "muz" şeklinde;
- omurga fıtığı.
Tarama birkaç düzlemde yapılmalıdır.
Bununla birlikte, görselleştirme aşağıdaki faktörler tarafından engellenebilir:
- hamile bir kadında obezite;
- karın ön duvarındaki sikatrisyel değişiklikler;
- çoklu hamilelik;
- muayene sırasında fetüsün uygunsuz pozisyonu;
- diğer nadir anomalilerin varlığı.
 Teşhisi netleştirmek için hamile bir kadına 20 haftalık gebelikten sonra bir MRG atanmalıdır. İlk trimesterde, embriyodaki hücre bölünmesi süreçlerini etkileyebileceği için bu yöntem kullanılmaz.
Teşhisi netleştirmek için hamile bir kadına 20 haftalık gebelikten sonra bir MRG atanmalıdır. İlk trimesterde, embriyodaki hücre bölünmesi süreçlerini etkileyebileceği için bu yöntem kullanılmaz.
Arnold-Chiari malformasyonlu yetişkinlerin ve çocukların tedavisi
Uzun yıllar değişmeden kalan belirgin klinik semptomları olmayan tip I sendromu olan hastalara, sağlıklarının dinamik olarak izlenmesi gösterilmektedir. Kontrol (röntgen muayenesi, ultrason veya MRI dahil) yılda en az bir kez gerçekleştirilir.
Diğer durumlarda ve hastanın durumu kötüleştiğinde cerrahi müdahale belirtilir.
konservatif terapi
Hafif bir ağrı sendromu olan hastalara aşağıdaki tabloda açıklanan semptomatik ajanlar reçete edilir.
| İlaç adı | Ana farmakolojik eylem | Günlük dozaj | Ortalama fiyat, ovmak. |
| gliatilin | Sinir uyarılarının iletimini iyileştirmek | 400 mg 3 kez | 710 |
| Alprazolam | Antikonvülsan, kas gevşetici | ¼ tablet 1 mg 3 kez | 850 |
| fluoksetin | antidepresan | sabah 2 mg | 100 |
| Diakarb | Diüretik (diüretik) | her biri 1 tablet | 280 |
| asparkam | Metabolik süreçlerin iyileştirilmesi | 1-2 tablet günde 3 defa | 65 |
| keton | Ağrı kesici | 1-2 kapsül 2-3 kez | 170 |
Cerrahi müdahale
Arnold-Chiari Sendromu, ana tedavisi cerrahi olan bir patolojidir. Omurga fıtığı varlığında yenidoğanda yaşamın ilk günlerinde yapılır.
Operasyon için endikasyonlar şunlardır:
- yaşam kalitesini ve hastanın durumunu önemli ölçüde bozan ciddi nörolojik semptomlar;
- siringomyeli ilerlemesi (omurilikte beyin omurilik sıvısı ile dolu boşlukların oluşumu);
- serebellumun sıkışması ve sıkışması nedeniyle şiddetli baş ağrısı.
Cerrahi müdahale aşağıdaki sırayla gerçekleştirilir:
- Ameliyat öncesi hazırlık (üst solunum yollarının obstrüktif patolojileri, akut ve kronik kardiyovasküler hastalıklar, hipertansiyon, aritmiler, sol ventrikül hipertrofisi ve diğer somatik patolojiler). Faaliyetlerin içeriği hastalığın türüne bağlıdır ve bireysel olarak belirlenir. Ameliyat sonrası komplikasyon riskini azaltmayı amaçlarlar.
- Hasta, oksipital bölgeye erişim sağlamak için yüzüstü, yan ya da yarı oturur pozisyonda yatırılır. Oturma pozisyonu aşırı kilolu hastalar için endikedir.
- Entübasyon (bir endotrakeal tüpün trakeaya yerleştirilmesi) bir hava yolu ve genel anestezi sağlamak için yapılır.
- Hastanın fizyolojik pozisyonunu sağlamak için silindirler kullanılır, kafa en az 3 noktada sabitlenir.
- Posterior median yaklaşımla serviko-oksipital bölgede cilt insizyonu yapılır.
- Kafatasının eksizyonu yapılır, omur kemerinin bir kısmı veya tamamı çıkarılır.
- Beynin araknoid zarında patolojik değişikliklerin ve zarın beyincik ile yapışmalarının varlığında, büyük oksipital sarnıcın açılması, yapışıklıkların eksizyonu yapılır.
- Beyincik C2 omurunun altına indiğinde rezeke edilir.
- Dördüncü ventrikülün genişlemesi ile şantlar implante edilir.
- Yapay malzeme kullanılan meninkslerin plastiği yapılır. Bazı durumlarda, sıkıştırmayı azaltmak için bir titanyum implant kullanılır.
- Yara, kas tabakası kullanılarak dikilir.
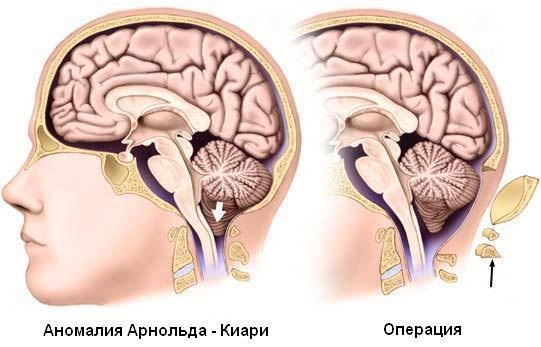
Ameliyat sonrası pozitif dinamikler hastaların yaklaşık% 70'inde gözlenir. Hastalığın süresi ile ters orantılıdır. Ameliyat edilenlerin bazılarında nöronlarda geri dönüşü olmayan değişiklikler nedeniyle yoktur. Çoğu zaman bu, klinik belirtilerin başlangıcından bu yana 2 yıldan fazla geçtiği durumlarda görülür. Hastaların %13-30'u tekrar ameliyat gerektirir.
Ameliyatın komplikasyonları şunlar olabilir:
- bulaşıcı süreçlerin gelişimi;
- beyin omurilik sıvısının çıkışı;
- zayıf yara iyileşmesi;
- servikal omurganın kararsızlığı;
- beyin sapını sıkıştıran bir hematom (kanama) oluşumu;
- aşırı kraniotomi nedeniyle beyincik prolapsusu;
- C1 vertebra yarım daire kırığı.
Ameliyat sonrası mortalite genellikle %2'yi geçmez.
Sonuçları nelerdir?
Zamanında tedavi eksikliği aşağıdaki sonuçlara yol açabilir:
- serebellar doku atrofisi;
- beyin nöronlarının yok edilmesi;
- koordinasyon ihlali, motor fonksiyonlar;
- görme ve işitme bozukluğu;
- felç;
- çocuklarda - bozulmuş büyüme ve zihinsel gelişim;
- akut solunum ve kalp yetmezliğine yol açan vejetatif bozukluklar;
- ölüm.
Arnold-Chiari sendromu (veya malformasyon) ciddi merkezi sinir sistemi bozukluklarına neden olur. Çoğu durumda bu hastalık kalıtsaldır, ancak hemen ortaya çıkmayabilir. Erken tanı yöntemleri hamilelik sırasında ultrason ve MRI içerir.
Ana tedavi yöntemi cerrahidir. Belirgin bir semptomatolojinin olmadığı durumlarda, beklenti taktikleri ve hastanın durumunun sistematik olarak izlenmesi tercih edilir.
Arnold-Chiari Sendromu Videoları
Arnold-Chiari Anomalisi: