 Les maladies du système nerveux sont fréquentes.
Les maladies du système nerveux sont fréquentes.
En général, ils se montrent névralgie et névrite, mais il existe aussi des formes de maladies héréditaires. Ceux-ci incluent l'ataxie spinocerebellar. Quelle est cette maladie?
Par « ataxie », on entend une condition neurologique dans lequel la cohérence est brisée mouvement conscient.
Contenu
- échec génique Type de trouble
- Manifestations
- premier type
- deuxième
- Maladie Machado-Joseph
- Type 5 et 6 - le résultat le plus favorable
- 7 visage de type cécité totale
- Comment établir le diagnostic?
- Cette maladie peut-elle être guérie?
- pronostic défavorable
échec Gene
La maladie est héréditaire et se développe en raison d'une défaillance génétique.
Ainsi, il peut se manifester sous deux formes - une violation du corps et de maintenir la stabilité dans l'espace( ataxie statique locomotrice à des lésions du vermis cérébelleux) ou avec des lésions et les abus hémisphères cérébelleux membres engagés mouvements actifs( forme dynamique de l'ataxie cérébelleuse).
La maladie est héritée par un type autosomique dominant. Les premiers signes apparaissent généralement sur l'âge de 30 ans .
clinique assez rare peut se développer avant, puis elle se manifeste le plus souvent en parallèle avec d'autres symptômes neurologiques.
En étudiant la structure génomique a été prouvé que dans le développement de l'ataxie spino premier rôle des gènes.
a prouvé qu'il ya un minimum de 13 gènes responsables du développement de l'ataxie, et plus précisément - un certain type.
est dans un chromosome particulier Chacun de ces gènes, et son échec en raison de et conduit à mutagenes le développement de la maladie.
On connaît actuellement 8 types de troubles. L'ataxie spinocérébelleuse de 1-13 types dépend de la lésion d'un gène spécifique. Le rôle principal est joué
en augmentant le nombre de triplets CAG( cytosine-adénine-guanine).Normalement, cette séquence nucléotidique codée par la formation de glutamine - un acide aminé nécessaire à la formation de liaisons entre les protéines du cerveau.
En raison de l'augmentation de sa quantité de chaînes de protéines d'allongement se produit, commencent ainsi à former des composants insolubles, à cause duquel l'interaction perturbée entre les parties du cerveau et la mort se produit un certain nombre de cellules nerveuses et favorise le développement de l'ataxie. Si l'on considère la substance 
cervelet, on peut voir que son hémisphère a subi des processus dégénératifs( ainsi que les principaux éléments impliqués dans la conduite de l'influx nerveux à la moelle épinière et le cervelet arrière( par exemple, d'olive)).
Parallèlement à cela, il y a démyélinisation des fibres nerveuses qui empêchent la propagation rapide de l'influx nerveux.
Manifestations troubles
Comment la maladie se manifeste?
Chacun des types d'ataxie est caractérisé par ses caractéristiques cliniques et d'âge. Type
premier premier type
se développe principalement dans les CAC personnes âgées de 30 à 40 ans. Une caractéristique de la maladie est qu'il peut se produire beaucoup plus tôt dans la prochaine génération que le précédent( phénomène que l'on appelle de l'anticipation).
premiers symptômes de la maladie sont généralement une certaine négligence de maladresses lors de mouvements rapides et intenses( par exemple, la course).
au fil du temps( en général quelques années), il y a d'autres symptômes - une violation de la stabilité dans la Romberg, dysarthrie, troubles de l'écriture particulière démarche ataxique.
Parallèlement à ces symptômes peuvent se développer tonique ou des convulsions cloniques, tendon augmenté ou l'apparition des réflexes pathologiques.
Dans les stades ultérieurs de la maladie peut se produire une démence, une altération de la phonation, de troubles pelviens.trouble du développement aigu du système nerveux périphérique, ce qui est le plus souvent causée d'une infection -
 polyradiculonévrite. Détails dans l'article.
polyradiculonévrite. Détails dans l'article. La leptospirose sévère chez l'homme est difficile à guérir. Mesures préventives de prévention, qui permettront d'éviter la maladie.
deuxième type
CAC deuxième type se produit presque aussi bien que le premier type et ataxie.
caractéristiques de la maladie est plus lent développement de saccades( mouvements concertés des globes oculaires) et ont exprimé l'anticipation, en particulier dans la ligne masculine.
Machado-Joseph maladie
maladie de Machado-Joseph( ataxie spinocérébelleuse type 3) a tout à fait un symptôme de polymorphisme prononcé.maladie
, en plus, cérébelleux et peut procéder à la clinique des troubles extrapyramidaux( tels que le parkinsonisme ou dystonie), amyotrophie et lésion des voies pyramidales.
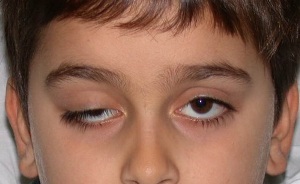
spécifique à MCA 3 symptômes sont ophtalmoplégie, phénomène « yeux bombé » , des contractions musculaires périoculaire rides MCA 4 en plus de flux principal troubles avec des symptômes de la neuropathie sensorielle. Type de
5 et 6 - le résultat le plus favorable
ataxie spinocérébelleuse de type 5 et 6 sont relativement favorables.
Ils développent à un rythme beaucoup plus tard l'âge, et les manifestations cliniques sont exprimés légèrement, ce qui permet aux patients de rester longtemps dans la société.
7 Type visage cécité totale
7 produit de type Ataxie normalement, avec la défaite de la rétine avec son détachement complet et le développement de la cécité.La déficience visuelle, souvent, est un signe avant-coureur des symptômes et peut considérablement surpasser les troubles de la coordination.
MCA 8 - MCA 13 est actuellement mal compris, comme sont extrêmement rares.
Comment établir un diagnostic?
méthodes d'imagerie préféré telles que CT ou IRM.
 Ils vous permettent de définir les zones de dégénérescence des fibres nerveuses démyélinisation des neurones du pont, l'expansion de tous les composants du système de circulation d'alcool dans le cerveau, les changements atrophiques du cortex cérébral.
Ils vous permettent de définir les zones de dégénérescence des fibres nerveuses démyélinisation des neurones du pont, l'expansion de tous les composants du système de circulation d'alcool dans le cerveau, les changements atrophiques du cortex cérébral.
Pour le diagnostic est également nécessaire d'exclure d'autres maladies qui se produisent avec une clinique similaire( par exemple une tumeur de la fosse cérébrale postérieure, la sclérose en plaques, hydrocéphalie, pathologie vasculaire cérébral, en particulier dans la colonne vertébrale et le département basilaire.
S'il est possible de confirmer le développement sur la base des données cliniquesataxie, il est beaucoup plus la définition du type de maladie est plus difficile( sinon des symptômes strictement spécifiques).
Le diagnostic moléculaire est recommandé de tenir ces personnes dans une famille où il y avait des cas de cette maladie.
Il est préférable d'effectuer des recherches prénatale à plus décider de la stratégie de traitement.
possible de guérir la maladie?
DansActuellement, il existe plusieurs opinions sur les tactiques de traitement de cette pathologie.
Les partisans de la première de l'avis que l'ataxie ne sont pas soumis à un traitement médicamenteux.
Dans ce cas, il est préférable d'effectuer un traitement d'entretien ralentit la progression de la maladie. Le complexe comprend une thérapie d'exercice obligatoire( formation vestibulaires), ainsi que de nombreuses méthodes de la vie sociale, la consommation et la réhabilitation du travail.
L'autre théorie suggère l'utilisation de certains médicaments, cependant, le traitement est purement symptomatique ou stimulant. 
Les injections de vitamines sont prescrites, ainsi que leur apport oral. En tant qu'élément d'une telle thérapie, il est possible de prescrire des anticonvulsivants( selon les indications).Traitement
ataxie spinocérébelleuse et certaines procédures de physiothérapie pris en charge( par exemple, électriques).
Prévisions décevantes
En ce qui concerne le pronostic de la maladie, les patients ataxiques vivent en moyenne entre 15 et 20 ans. Avec des soins appropriés, ainsi qu'une maladie diagnostiquée en temps opportun et l'utilisation d'un traitement d'entretien, il est possible de prolonger la vie de 7 à 10 ans.
Les patients ayant une ataxie de 5 ou 6 types peuvent vivre normalement un peu plus longtemps( la durée de vie ne change généralement pas beaucoup).
Étant donné que la maladie est mal traitée, il est nécessaire d'améliorer les méthodes de diagnostic disponibles, ainsi que d'essayer de découvrir et de mettre en œuvre de nouvelles tactiques thérapeutiques.