 Doenças degenerativas e distróficas do sistema nervoso com lesão predominante de nervos periféricos e fibras musculares ocupam uma grande parte da estrutura da patologia hereditária humana.
Doenças degenerativas e distróficas do sistema nervoso com lesão predominante de nervos periféricos e fibras musculares ocupam uma grande parte da estrutura da patologia hereditária humana.
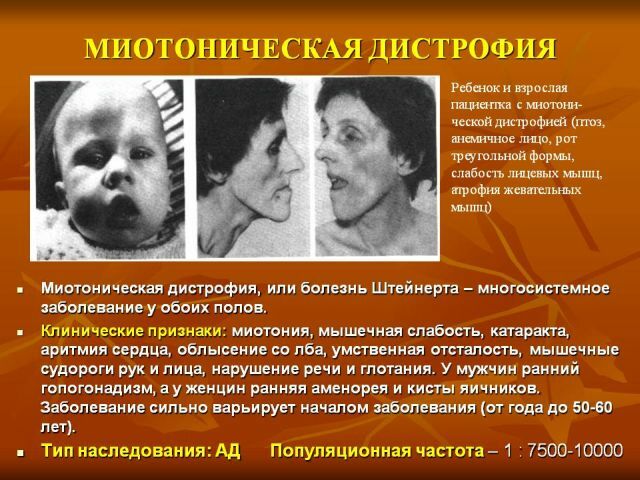
Um representante típico é a distrofia miotônica( ou miotonia distrófica), descrita no início do século passado por vários autores e denominada doença Rossolimo-Steinert-Kurshmana.
Esta doença é a doença mais famosa da categoria de miotonia e a forma mais comum de distrofia muscular em adultos. Qual é esta doença e como lutar contra isso?
Descobrindo e a essência da doença
Rossolimo, Steinert e Kurshman estudaram uma doença que é uma patologia genética com um tipo de herança autossômica dominante. Isso significa que um dos pais possui um gene mutante, nascem crianças doentes com uma probabilidade de 50%.A doença tem a natureza de uma aflição familiar e é transmitida para as gerações subsequentes em escala vertical.
Filhos e filhas em tais famílias estão doentes com a mesma freqüência, aproximadamente 3 a 5 pessoas por 100 mil habitantes. A idade de início da doença, bem como a gravidade dos sintomas, são marcadamente variáveis.
As formas neonatais e tardias precoce são descritas, mas na maioria das vezes a doença faz sua estréia no segundo, raramente - na terceira década de vida.  Observa-se que a transmissão da doença à criança da mãe é mais prognosticamente desfavorável do que a do pai.
Observa-se que a transmissão da doença à criança da mãe é mais prognosticamente desfavorável do que a do pai.
No coração da doença é um gene de defeito de 19 pares de cromossomos, que é responsável pela síntese da enzima miotonina-proteína quinase. Esta proteína está normalmente presente não apenas na musculatura esquelética, mas também nas células do miocárdio e do sistema nervoso central.
É por isso que, para a miotonia distrófica, caracterizou manifestações polissistêmicas com a derrota de vários órgãos e sistemas. A incompletude da miotonina-proteína quinase leva à aparência de espasmos musculares, juntamente com mudanças atróficas na musculatura da cabeça, região cervical e membros.
Existe uma combinação de hipertrofia de algumas fibras musculares com atrofia de outros e sua substituição por tecidos gordurosos ou conectivos.
Manifestações clínicas de
Devido à variação no início da doença, as seguintes formas relacionadas à idade são distinguidas na clínica:
- , forma congênita de - a manifestação da doença começa imediatamente após o nascimento da criança;
- versão para jovens de - debut de miotonia na idade de um ano para a puberdade;
- , forma clássica de - o início das manifestações clínicas cai na segunda e terceira dúzia de vidas;
- é a versão mínima do - a manifestação cai nos termos tardios - a sexta dozinha da vida.
Caracteristicamente, quanto mais tarde manifesta a doença, mais favorável é o curso e melhor o prognóstico. A forma clássica da doença de Steinert é mais comum, para a qual os seguintes sintomas clínicos são típicos:
- Myotonia - manifestada por espasmos dos músculos mastigatórios e flexores das mãos, as alterações atróficas em diferentes grupos musculares
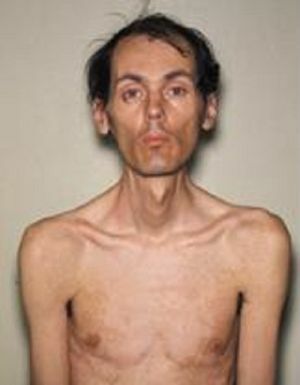 são características. Gradualmente, a desvanecimento das manifestações miotônicas e a progressão da distrofia muscular ocorre externamente e é expressa em uma máscara triste do rosto e ausência de expressão facial. Perigoso é a paresia dos músculos da laringe com distúrbios de deglutição, bem como a fraqueza dos músculos respiratórios, pelo que há possíveis ataques de parar de respirar em um sonho, o desenvolvimento de pneumonia.
são características. Gradualmente, a desvanecimento das manifestações miotônicas e a progressão da distrofia muscular ocorre externamente e é expressa em uma máscara triste do rosto e ausência de expressão facial. Perigoso é a paresia dos músculos da laringe com distúrbios de deglutição, bem como a fraqueza dos músculos respiratórios, pelo que há possíveis ataques de parar de respirar em um sonho, o desenvolvimento de pneumonia. - Distúrbios cardiovasculares - distúrbios do ritmo cardíaco, alterações hipertróficas do ventrículo esquerdo, detectadas no ECG, insuficiência cardíaca congestiva.
- Doenças endócrinas ( principalmente afetadas pela função sexual) - reduzindo o tamanho dos órgãos genitais, reduzindo o desejo sexual, nas mulheres - distúrbios menstruais, obesidade.
- Alterações gerais na natureza distrófica de - secura e pigmentação da pele, perda parcial ou total de cabelo e dentes, catarata precoce.
- Distúrbios do SNC - fadiga, distúrbios do sono, apatia, perda de inteligência.
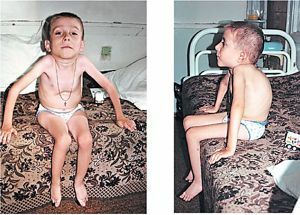
Separadamente, vale a pena observar as manifestações clínicas características da forma congênita de miopatia distrófica:
- diminui nos movimentos fetais ativos no útero, detectados durante o ultra-som;
- durante o período do recém-nascido - letargia, hipotensão generalizada, especialmente na mastigação, músculos faciais dos globos oculares;Preservação
- e mesmo aumento de reflexos tendinosos;Problemas de alimentação com
- , dificuldade respiratória como síndrome de dificuldade respiratória;
- atraso do desenvolvimento físico e neuropsíquico, sinais de oligoprenia;
- progressão rápida da doença, alto risco de morte súbita.critérios
diagnóstico
suspeita de doença Rossolimo-Steinert - Kurshmana pode ocorrer no médico se o paciente tem uma combinação de distrofia miotônica e alterações nos músculos no fundo da perda de inteligência e presença de doenças cardiovasculares e endócrino.
Polysystemic quase sempre indica a natureza genética da doença. Tais pacientes estão sujeitos a análise de DNA e análises genealogicas para confirmar a herança de patologia autossômica dominante. Como métodos informativos de pesquisa, eletrocardiografia, eletroneuromiografia, testes hormonais são utilizados.
Devido à versatilidade das manifestações clínicas do processo de diagnóstico são geralmente especialistas envolvidos de diferentes ramos da medicina - a genética, cardiologia, endocrinologia, ginecologia, andrologia, neurologia.
O diagnóstico diferencial é feito entre miotonia distrófica e outros tipos de doenças semelhantes. Ao contrário do resto, a atrofia muscular é característica da doença de Rossolimo. Muitas vezes, para confirmar o diagnóstico, você precisa recorrer a uma biópsia para determinar o nível de proteína muscular, que é aumentada nos tecidos dessa patologia.
O diagnóstico pré-natal também é realizado utilizando o método de pesquisa de líquido amniótico.doença genética
Assistência Médica
não pode ser curada completamente, de modo que o objetivo do tratamento na doença Rossolimo-Steinert-Kurshmana é o alívio dos sintomas, melhora na condição geral e adaptação social dos pacientes.
princípios de tratamento são os seguintes: sais de
- dieta baixa potássio( maçãs, espargos, couve, Pepino, uva, verdes, milho, bagas, rabanetes, tangerinas, toranjas
 , cebolas, cenouras, berinjela, ervilha);Supressão
, cebolas, cenouras, berinjela, ervilha);Supressão - do superenfriamento para evitar espasmos;
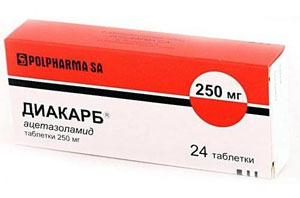
- uso de drogas quinina para estabilizar as membranas celulares de tais medicamentos como Difenin, procainamida, Diakarb - para aliviar os espasmos musculares e rigidez reduzida, convulsões, diminuir a pressão intracraniana;
- uso de esteróides anabolizantes ( Metanandrostenolona, Retabolil, Nerabol), vitaminas B, ATP para estimular a massa muscular;
- LFK, massagem, eletromiostimulação, adaptações ortopédicas .
As medidas listadas dão um bom efeito positivo na forma clássica e congênita da doença. Livrar completamente o paciente da miotonia distrófica, eles não podem, mas prolongar sua vida e melhorar sua qualidade são capazes.
O prognóstico é pior na forma congênita - a letalidade é alta, as crianças podem não viver até 3 anos. A versão juvenil da miotonia prossegue suficiente 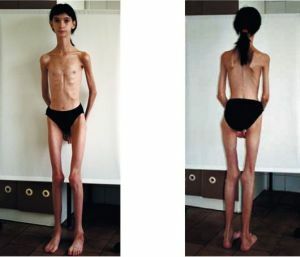 é difícil e pode levar já em anos jovens a deficiência e deficiência antecipada.
é difícil e pode levar já em anos jovens a deficiência e deficiência antecipada.
No caso da forma clássica, a doença pode levar muito tempo no decurso de medidas oportunas de tratamento e correção. O prognóstico mais favorável nas formas de início tardio da doença. Medidas preventivas
são reduzidos ao fato de que mulheres de famílias desfavorecidas com história na fase de planejamento da gravidez é necessário ser rastreados para a presença de genes anormais responsáveis pelo desenvolvimento de distrofia muscular. Também é aconselhável fazer isso se a patologia dos parentes do pai estiver presente.
As oportunidades para o nascimento de crianças devem ser decididas individualmente em cada caso por geneticistas após a consulta.