
מחלות תורשתיות של בני אדם נגרמת על ידי מוטציה גנטית המובילה להופעת תכונות חדשות וספציפיות. בחלק מהפתולוגיות, סימפטומים ספציפיים מופיעים מיד לאחר לידתו של ילד, עבור אחרים, יש קורס סמוי המתבטא רק כאשר מגיעים לגיל או מצב מסוים.
יש להבדיל בין רשימת המחלות התורשתיות לבין פתולוגיות מולדות ומשפחתיות. הראשון יכול להיגרם הן על ידי גורמים שליליים גנטיים והן מכניים, כימיים, ביולוגיים, וסיבה תכופה לאחרונים היא הנטייה המשפחתית של האדם.
בהתאם לסוג ההתרחשות, ניתן לחלק מחלות תורשתיות ל:
- גן הקשור לפגיעה במבנה או בחלק משרשרת ה- DNA;
- כרומוזומלי, הנגרם על ידי חריגות במספר או פגם במבנה הכרומוזומים;
- רב -פקטוריאלי (או מחלות בעלות נטייה תורשתית), שינה בגוף האדם עד לנקודה מסוימת, מעוררת על ידי גורמים חיצוניים.
הקלט תוכן:
-
1 גֵנֵטִי
- 1.1 תורשה אוטוזומלית דומיננטית
- 1.2 קשור לשידור אוטוזומלי רצסיבי
- 1.3 ירושה של כרומוזום X
- 1.4 ירושה של כרומוזום Y
-
2 כרומוזומלי
- 2.1 נגרם על ידי טריזומיה
- 2.2 מונוסומיה
- 2.3 נגרם כתוצאה מאובדן הכרומוזומים
- 3 רב -פקטוריאלי
- 4 סרטונים על מחלות תורשתיות
גֵנֵטִי
מחלות אנוש תורשתיות (רשימה של מחלות מובאת להלן במאמר) הקשורות להפרה של מבנה שרשראות ה- DNA ו פגיעה בגנים האחראים להעברת מידע תורשתי לילד משני ההורים שייכים לקבוצת הגנים פתולוגיות.
לרוב, המונח "גן" פירושו פתולוגיות מונוגניות הנגרמות על ידי מוטציות או חוסר גנים בודדים ועברו בירושה בהתאם לחוק מנדל (אוטוסומלי או מקושר לכרומוזום X) דרכים).
תורשה אוטוזומלית דומיננטית
מחלות אנוש תורשתיות (רשימת הפתולוגיות התורשתיות נערכה על ידי גנטיקאים מובילים) המתרחשות בכל דור הן פתולוגיות, מועבר בדרך של ירושה אוטוזומלית דומיננטית, אם אדם יורש כרומוזום תקין ואחד "משתנה", הנמצא בדומיננטי (המיוחס) מַצָב.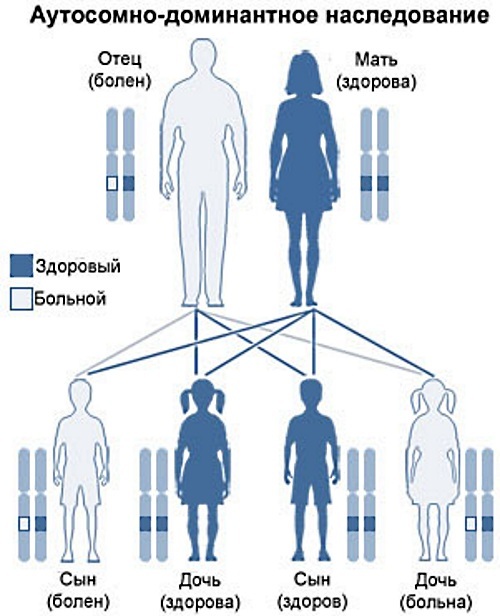
| שם רפואי | שלטים | סיבות להופעה | התפתחות | את מי אפשר להעביר |
| תסמונת מרפן |
עבור אנשים הסובלים מפתולוגיה, זה אופייני:
|
תת -התפתחות מערכתית של רקמת חיבור הנגרמת מפגם בקולגן ומלווה בפגיעה במבנה העיניים, מערכת הלב וכלי הדם או מערכת השלד והשרירים. | ילד עם פתולוגיות מאופיין בהתפתחות של מחלות לב מולדות, פתולוגיות של אברי הראייה, פגיעה במערכת הסימפונות והעצבים המובילה למוות עד גיל 30. | גם בנים וגם בנות סובלים מפתולוגיות בתדירות שווה. מספר מקרי הפתולוגיה הוא 1 מכל 10,000-20,000 אנשים. |
| נוירופיברומטוזיס של רקלינגהאוזן |
הסימפטומטולוגיה של המחלה משתנה בהתאם למיקום הגידולים, ובשלב הראשון היא מתבטאת בהופעת כתמי גיל חלב על העור:
|
פתולוגיה המאופיינת בהתפתחות גידולים (נוירופיברומט) במערכת העצבים וברקמות היקפיות. | עם המשך הקורס מתרחשת היווצרות של גושים תת עוריים מרובים המופיעים על תא המטען, הצוואר, הראש והגפיים. התפתחות הנוירופיברומות בעצבי הראייה והשמיעה מלווה בעיוורון מוחלט ואובדן שמיעה. | הפתולוגיה מאובחנת בגיל 3-16 שנים, הן אצל בנים והן בנות. התפשטות המחלה היא מקרה אחד לכל 4000-5000 לידות. |
| תסמונת אהלרס-דנלוס |
תסמיני המחלה הם מערכתיים, כלליים, המתבטאים:
|
דיספלסיה תורשתית, רקמת חיבור, הנגרמת על ידי ייצור לא מספיק של חלבון עצם (קולגן). | בשל העובדה כי הטיפול במחלה לא פותח, המטופל זקוק להגבלה קפדנית של הגוף עומס, שכן מוות יכול להתרחש עקב קרע בכלי הדם ובפנים מְדַמֵם. | משפיע על ילדים משני המינים, ומופיע אצל 1 מכל 5,000 ילודים. |
| Osteogenesis imperfecta |
מחלות של מערכת השרירים והשלד מתבטאות ב:
|
מוביל לבעיות בהיווצרות עצם ונטייה לשברים תכופים. | ילדים עם התסמונת מתים במשך 2-3 החודשים הראשונים לחיים עקב שברים מרובים או סיבוכים ספטיים. | המחלה מאובחנת אצל תינוקות משני המינים בתדירות של 1 מכל 10 אלף. יילודים. |
| Pseudohypoparathyroidism או מחלת Albright |
ניוון תורשתי של רקמת שריר -שלד, המלווה בעלייה בתכולת הסידן בדם, מאופיין בהפרעות מטבוליות, התפתחות פיזית ונפשית מאוחרת. סימפטומים של פסאודוהיפופאראתירואידיזם כוללים:
|
הופעת המחלה נובעת מהתנגדות השלד והכליות לפעולה של הורמון הפאתירואיד הנגרם כתוצאה מפגם במבנה ממברנות התא. | בעת ביצוע טיפול החלפת הורמונים, קיימת סבירות גבוהה להפחית את ביטוי המחלה ולנרמל את המצב האנושי. | פתולוגיה תורשתית נדירה שאובחנה ב -300 חולים בלבד. |
קשור לשידור אוטוזומלי רצסיבי
העברה אוטוזומלית רצסיבית מתאפיינת בהכללת הדנ"א של הילד של גנים ממוטציה מזווגים הנמצאים בגנוטיפים של שני ההורים.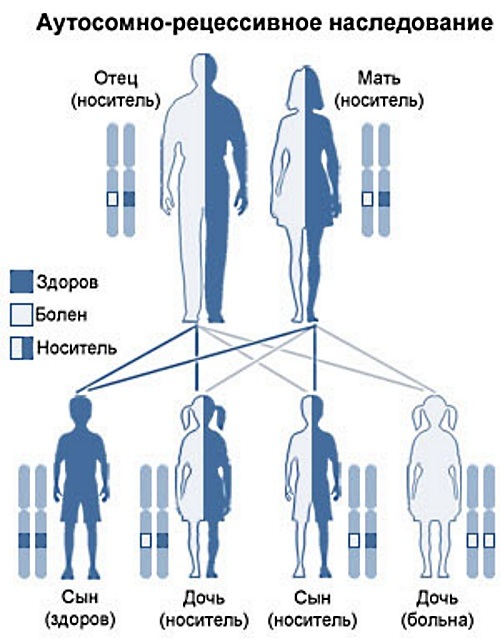
| שם רפואי | שלטים | סיבות להופעה | התפתחות | את מי אפשר להעביר |
| פנילקטונוריה | הפתולוגיה מלווה בהקאות קשות, עייפות או היפראקטיביות, ריח עובש משתן ועור, פגיעה בהתפתחות פסיכומוטורית. | הפרעה גנטית של חילוף החומרים של חומצות אמינו, מעוררת מחסור באנזים בכבד. | הסימנים הראשונים למחלה מופיעים בגיל 2-6 חודשים לחיים, מרגע שהחלבון הראשון נכנס לגוף והופך מיד להקאה מתמשכת. עד שישה חודשים המחלה מתקדמת ומעוררת עיכוב בהתפתחות הפסיכו -מוטורית. | מופיע בילדים משני המינים בתדירות של מקרה אחד לכל 10,000 תינוקות. |
| גלקטוזמיה |
מעורר:
|
הפרת מטבוליזם אנזימטי ופחמימתי הקשורה לתפקוד לקוי של מטבוליזם הלקטוז. | התפתחות המחלה מתרחשת מיד לאחר הלידה, המתבטאת בצואה מימית והקאות מתמשכות, ועל ידי 2-3 חודשים של חיים, הילד חווה עיוות ושחמת הכבד, דלקת כבד, כבד חריף כישלון. | מחלה גנטית נדירה, מאובחנת במקרה אחד לכל 10,000 - 50,000 תינוקות. |
| קַשׂקֶשֶׂת | הוא מלווה בהפרה של קרטיניזציה של רקמות, המתבטאת בצורה של היווצרות קשקשים על העור בצורה של קשקשי דגים. | מחלת עור תורשתית. תורש רק בכרומוזום X. | הפרוגנוזה של המחלה אינה חיובית והיא כרוכה בתוספת של פתולוגיות מערכתיות המעוררות מוות מוקדם. | לרוב מאובחן אצל גברים בגיל 3 שנים. |
| פרוג'ריה |
המחלה מתבטאת בילד:
|
פתולוגיה גנטית נדירה יחסית הקשורה להזדקנות מוקדמת של איברים פנימיים, המובילה לשינויים מקבילים בגוף. פתולוגיות גנטיות מועברות רק בנוכחות גנים מוטציות בכרומוזומי המין. | אורך חייו של ילד חולה הוא בין 3 ל -13 שנים. | מחלות כאלה באות לידי ביטוי בילדים שנולדו מנשא א -סימפטומטי של גנים פגומים בכרומוזומי Y או X. |
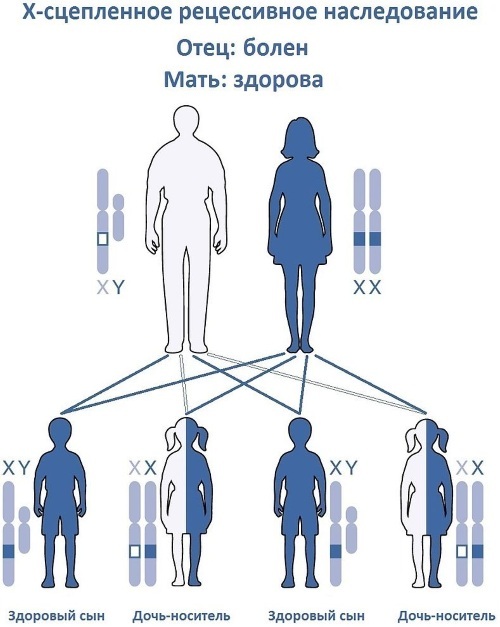
ירושה של כרומוזום X
תורשה רצסיבית צמודה ל- X מתרחשת רק אם הגן הפגום (מוטציה) נמצא בכרומוזום המין של אמו של המארח והוא הומוזיגוט או הטרוזיגוט. פתולוגיות כאלה מועברות מנשאים בריאים רק לבנים שנולדו על ידם.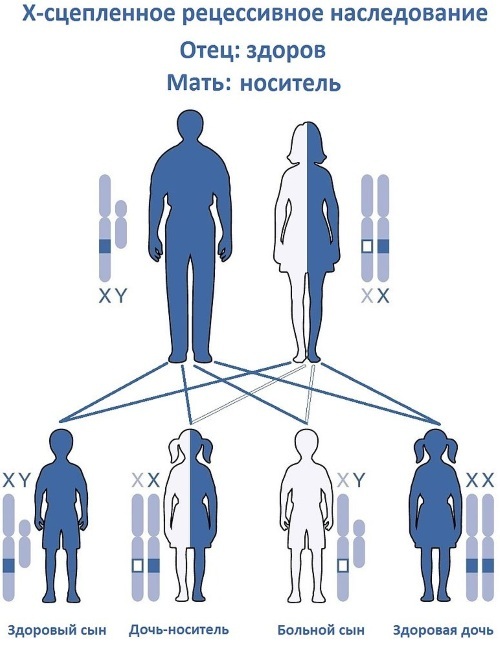
| שם רפואי | שלטים | סיבות להופעה | התפתחות | התפשטות הפתולוגיה |
| דַמֶמֶת |
הביטויים הספציפיים ביותר של המופיליה הם הנטייה לדימום פנימי וחיצוני:
|
פתולוגיה תורשתית של המוסטזיס, המבוססת על הפרה של תהליך קרישת הדם. | עם טיפול חלופי ממושך, מצבו של המטופל מנורמל. מחלה קלה אינה משפיעה על תוחלת החיים. במקרים חמורים כל פציעה חמורה והליכים כירורגיים עלולים להיות קטלניים. | מקרה אחד לכל 10,000-50,000 זכרים. |
| פוספט-סוכרת |
הסימפטומטולוגיה של המחלה מורכבת מ:
צורות מסוימות של פתולוגיה מתאפיינות גם בפגיעה בספיגה של סידן ותרכובותיו על ידי הגוף, מה שמוביל לעלייה במדד ההיפוגליקמי ובפתולוגיה של בלוטות התריס. |
מצב פתולוגי המאופיין בבעיות בספיגת הזרחן ומוביל להפרה של מבנה העצם ורקמת החיבור. | עם טיפול תחליפי, הפרוגנוזה של המחלה היא חיובית, אך עבור המטופל לאורך חייו, הצורך בשימוש קבוע בוויטמינים D, זרחן וסידן יישאר. עיוות מולד שלד יכול גם להחמיר את איכות החיים. | אבחון הפתולוגיה הוא 1: 20,000 בנים שזה עתה נולדו. |
| ניוון שרירים של דושן | מתחיל להתבטא בגיל 3-5 שנים עם חולשת שרירים מתפשטת במהירות ומתקדמת. | השם השני של המחלה הוא מיופתיה. הפתולוגיה עוברת בירושה לאורך כרומוזום X. | בתחילה, המחלה מתפשטת לשרירי חגורת האגן והירכיים, ולאחר מכן משפיעה על הכתפיים והגב, ומעוררת את המראה של חוסר תנועה מוחלט. במקביל מתרחשות עיוות שלד ופגיעה במערכת הלב וכלי הדם. | לרוב בנים סובלים מפתולוגיה (מקרה אחד מכל 4 אלף). |
| תסמונת האנטר |
מוביל להופעה של:
|
פתולוגיה מטבולית תורשתית הקשורה לייצור אנזימי אידורסולפאז על ידי הגוף והצטברות של מוקופוליסכרידים ברקמות. תורשה מתרחשת בכרומוזום מין X. | התסמינים מתפתחים בין הגילאים 1 עד 3 שנים. תוחלת החיים היא כ- 50-60 שנים. | שיעור הילודה של ילדים הסובלים מתסמונת האנטר הוא כ -1 מתוך 100,000 עד 150,000 יילודים. |
| מחלת פאברי |
המחלה מסודרת:
|
פתולוגיה הנגרמת על ידי מוטציה של הגן נובעת מחוסר פעילות או היעדרות מוחלטת של האנזים גלקטוסידאז בחילוף החומרים של השומנים. |
הוא מופעל על ידי 6-12 או 30 שנים. עם טיפול בזמן, הסימפטומים נעצרים. תוחלת החיים של המטופל היא כ- 40-50 שנה. |
השכיחות הגבוהה ביותר של המחלה נמצאת בארצות הברית. התפשטות הפתולוגיה היא מקרה אחד לכל 40-120 אלף. יילודים. |
ירושה של כרומוזום Y
תורשה בכרומוזום Y מתרחשת לעתים רחוקות למדי ומתרחשת רק מהאב ומועברת אך ורק לזכר. סימנים לפתולוגיות כאלה נמצאים בכל דור המשפחה.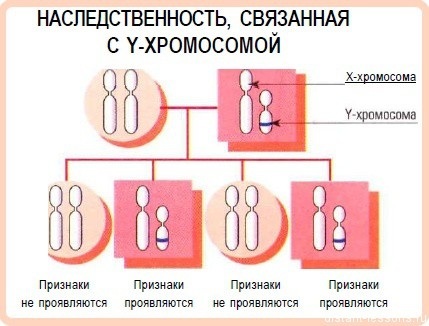
| שם רפואי | שלטים | סיבות להופעה | התפתחות | כמה פעמים זה קורה |
| תת פעילות של בלוטת התריס של האפרכסת | הוא מלווה במראה השיער וצמיחתם העודפת באזור האפרכוסים. | נגרמות על ידי מוטציות גנטיות שמשנות את מבנה תאי האפיתל, והופכות את המבנה שלהן לתאים אפידרמיסיים. | הוא מתבטא בילדות המוקדמת בצמיחת קו השיער במקומות לא סטנדרטיים. | בנים בלבד. |
כרומוזומלי
מחלות אנוש תורשתיות (רשימת המחלות המועברות גנטית כוללת, בנוסף לחריגות גנים ופתולוגיות, הקשור לשינוי בשרשרת ה- DNA של קבוצת כרומוזומים) יכול להיגרם על ידי מוטציות המשפיעות לא על גנים שלמים, אלא רק על אינדיבידואלים כרומוזומים.
תכונה ייחודית של קבוצה זו של פתולוגיות תורשתיות היא הכללת כרומוזומים חדשים בשרשרת ה- DNA או אי הכללת כרומוזומים שנפלו, כמו גם שינוי או נזק מהמבנה.
מוטציות כרומוזומליות מתרחשות עקב תקלה בגנוטיפ ההורי ונחשבות לאחת הסיבות העיקריות המעוררות הופעת הפלות ספונטניות או מוות תוך רחמי של העובר. לא יותר מ 3-5% מהם עוברים מדור לדור.
נגרם על ידי טריזומיה
טריזומיה היא מוטציה גנטית, וכתוצאה מכך מופיע כרומוזום נוסף נוסף בגנוטיפ האנושי, המוביל להתפתחות של פתולוגיות תורשתיות קשות.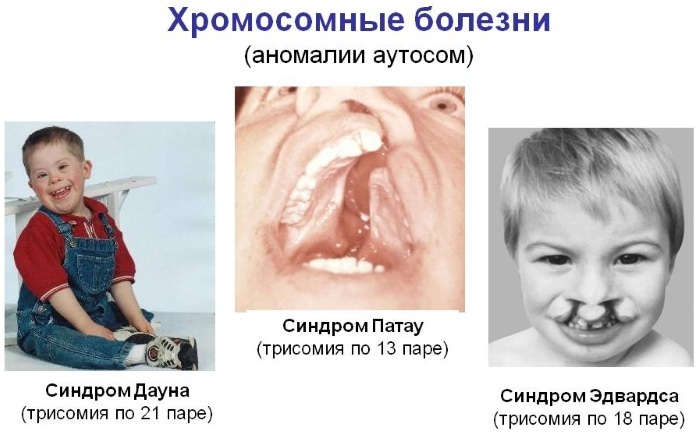
| שם רפואי | שלטים | סיבות להופעה | התפתחות | את מי אפשר להעביר |
| תסמונת דאון |
הסימנים האופייניים לפתולוגיה הם:
|
אנומליה המתבטאת בעלייה (שילוש) של העתקים של חומר גנטי בכרומוזום ה -21. | הפתולוגיה המתרחשת בתקופה של התפתחות תוך רחמית יכולה להסתיים בהפלה ספונטנית או ללוות את לידתו של ילד עם תכונות של מבנה הגוף והתפתחותו. רוב הילדים הסובלים מהתסמונת מצליחים להקנות כישורי בית ותקשורת מינימליים. חולים בוגרים עם התסמונת יכולים לנהל חיים עצמאיים. | הפתולוגיה, המופיעה בכ 1-2 חולים לכל 300-400 איש, נחשבת לחריגה הכרומוזומלית השכיחה ביותר. |
| תסמונת פטאו |
תינוקות שנולדו עם תסמונת פטאו שונים:
|
עלייה במספר הכרום בזוג ה -13. | לרוב, הפתולוגיה מאובחנת עם לידת מת של ילד. תוצאה קטלנית יכולה להתרחש גם במהלך השנה הראשונה לחייו של התינוק. | שיעור הילודה של תינוקות משני המינים הוא 1: 7000-10000. |
| תסמונת אדוארדס |
זה בא לידי ביטוי במראה:
|
המחלה נגרמת על ידי טריזומיה של הזוג ה -18. | רוב הילדים הסובלים מפתולוגיה זו מתים לפני הגעתם לשלושה חודשים עקב דום נשימתי או הפרעות בקצב הלב. |
נראה לרוב אצל תינוקות שנולדו לאמהות מעל גיל 45 שיעור הילודה של ילדים הוא 1: 5000-7000. רוב הפתולוגיות החולות הן בנות. |
| תסמונת טריזומיה X | סטייה קלה בהתפתחות הפסיכו-רגשית, המאופיינת בירידה קלה באינטליגנציה ובנטייה של האדם להופעת סכיזופרניה או הפרעה דו קוטבית. | הוא נגרם מהיווצרותם של שלושה כרומוזומים בזוג ה -47, 4 בכ -48 או 5 בכרומוזום ה -49. | רוב מקרי הפתולוגיה נותרים ללא גילוי, ולתינוקות אין פגמים חיצוניים ומנהלים חיים תקינים. | פתולוגיה מופיעה בכ -1 מכל 1000 ילדות שזה עתה נולדו. |
| תסמונת קלינפלטר |
בא לידי ביטוי בהתפתחות: אי פוריות גברית;
|
פתולוגיה תורשתית הקשורה לעלייה בגנוטיפ הגברי (זוג 47, 48, 49) של מספר הכרומוזומים הנשיים. | עם הזמן, סכיזופרניה ודמנציה מתפתחים, ומלווה גם באי פוריות. | זה מופיע אצל 1.5 מתוך 1000 בנים. |
| תסמונת ג'ייקובס | עשוי להיות מלווה בהתקפי תוקפנות בלתי מוסברים, עם חולשה קלה בו זמנית ופיגור פסיכולוגי. | זה קשור לפער במספר כרומוזומי X במהלך הזרע ובעלייה בקריוטיפ בזוג ה -47. | לילדים עם התסמונת אין שום מוזרויות התפתחותיות, אך הם תמיד גדלים מהר הרבה יותר מבני גילם. | היא מופיעה אצל גברים גבוהים עם פנוטיפ רגיל יחסית. התרחשה אצל 1 מתוך 1000, היא אובחנה לרוב בפושעים תוקפניים. |
מונוסומיה
מונוסומיה של כרומוזומי המין מלווה בהכפלה או שליטה מלאה של כרומוזום מין X, המוביל לחריגות התפתחותיות.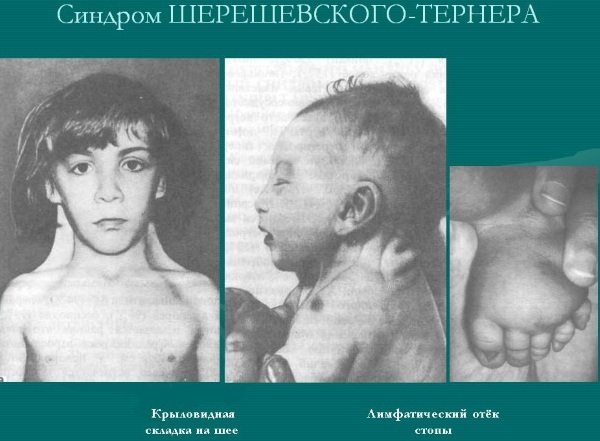
| שם רפואי | שלטים | סיבות להופעה | התפתחות | את מי אפשר להעביר |
| תסמונת שרבסקי-טרנר |
המחלה מתבטאת:
|
אנומליה גנטית נקבה, המלווה באי התאמה במספר כרומוזומי המין (היעדר אחד מכרומוזומי ה- X שלהם). | אינו משפיע על תוחלת החיים, אך מתבטא בהתפתחות אי פוריות. | זה קורה אצל 1 מתוך 3000 ילדות שזה עתה נולדו. |
נגרם כתוצאה מאובדן הכרומוזומים
מחלות תורשתיות של בני אדם המתפתחות כתוצאה ממחיקת מיקרו של כרומוזומים נגרמות על ידי אובדן או אובדן חלק מהמבנה הגנטי (זרוע כרומוזומלית) המוביל להתפתחות גנטית רצינית חריגות.
ברשימת הפתולוגיות כאלה:
| שם רפואי | שלטים | סיבות להופעה | התפתחות | את מי אפשר להעביר |
| תסמונת וויליאמס |
זה בא לידי ביטוי בהתפתחות של פיגור שכלי וחריגות רבות של התפתחות גופנית. סימנים אופייניים לפתולוגיה הם:
|
פתולוגיה גנטית הנגרמת כתוצאה מאובדן או הרס של חלק מהכרומוזום השביעי. סבורים כי תסמונת פני השדיים נגרמת כתוצאה ממוטציה חדשה בגנוטיפ ההורי. | מוביל להתפתחות של מחלות לב, אך עם חומרה קלה והקלה בסימפטום, החולה יכול לחיות עד לגיל מבוגר. | זה משפיע על בנים ובנות עם אותה תדירות, ומופיע אצל 1 או 20 אלף. מקרים. |
| תסמונת אנג'למן |
לאנשים הסובלים מפתולוגיה זו, המאפיינים הבאים הם:
|
אובדן חלק מהכרומוזום ה -15 גורם לפתולוגיות התפתחותיות חמורות. | עם הזמן הסימפטומים הופכים פחות בולטים וכישורי השליטה העצמית משתפרים. | זה קורה במקרה אחד מתוך 10-20 אלף. יילודים. לרוב המחלה מאובחנת אצל בנים. |
| תסמונת פראדר ווילי |
מלווה ב:
יש גם מבנה מעוות של הפנים (אף שטוח ואוזניים, חך גבוה, שפה עליונה קצרה) והתפתחות לא תקינה של איברי המין. בגיל ההתבגרות גם סוכרת מצטרפת לפתולוגיה. |
הרס שני החלקים (אימהית ואבהית) בכרומוזום 15 תורם להתפתחות חריגה גנטית חמורה. | זה מעורר התפתחות של אי ספיקה ריאתית, שהיא לעתים קרובות הסיבה למוות בגיל 4-5 שנים. במקרה של הקלה בזמן בסימפטומים תוחלת החיים היא 60-70 שנים. | שכיחות המחלה היא 1: 15,000 - 1: 25,000. אין הבדלים בין המינים. |
| תסמונת צרחות חתולים |
ילדים הסובלים מפתולוגיה זו מאופיינים בנוכחות:
תכונות המחלה נחשבות גם לאידיופתיה עמוקה, היפראקטיביות ונטייה לתוקפנות בלתי מבוקרת. |
היעדר קטע של הכרומוזום החמישי, שקיבל את שמו מצלילי הצעקות שהשמיעו תינוקות, הדומה במידה רבה למיאו של חתול. | הוא מלווה בהופעת פיגור רציני ברמת ההתפתחות הפסיכו -רגשית. | היא נחשבת לאחת הפתולוגיות הנדירות המתרחשות באחד מכל 45 אלף איש. יילודים. |
רב -פקטוריאלי
מחלות אנוש תורשתיות (רשימת הפתולוגיות בעלות נטייה גנטית מכילה פתולוגיות מסכנות חיים, ככלל, מתבטאות בבגרות) היא קבוצה של מחלות רב -גורמיות המשלבות שינוי בגנוטיפ האנושי והשפעת גורמים חיצוניים גורמים.
מועברות יחד עם שרשרת ה- DNA, מחלות כאלה נשארות לעיתים קרובות מבלי לשים לב לגוף עד לרגע המראה גורמים סביבתיים המעוררים את התפתחות הפתולוגיה, כולל:
- לעשן;
- אכילת הרבה שומנים מן החי;
- שמירה על אורח חיים בישיבה;
- הַשׁמָנָה;
- מתח עצבי חזק ומתמיד;
- לאכול הרבה סוכר ומלח.
| שם רפואי | שלטים | סיבות להופעה | התפתחות | את מי אפשר להעביר |
| טרשת עורקים |
כהופעות של המחלה, לרוב ניתן לצפות בהתקפי אנגינה פקטוריס, מה שמוביל להופעת:
|
פגיעה מערכתית בעורקים, מלווה בהופעת מרבצי כולסטרול גדולים בציפוי הפנימי של כלי הדם, המובילים להיצרות לומן ופגיעה באספקת הדם לשריר הלב. | מוביל להתפתחות של אי ספיקת לב וכלי דם חריפה או כרונית. | לרוב זה משפיע על גברים בגילאי 40-45 שנים. |
| מחלה היפרטונית |
התסמינים העיקריים של יתר לחץ דם נחשבים:
|
פתולוגיה של מערכת הלב וכלי הדם הנגרמת כתוצאה מתקלה במרכז ויסות כלי הדם ו מנגנוני כליות ומובילים לעלייה בפרמטרים הפיזיים של לחץ הדם, שינויים בעבודת הלב, מערכת העצבים המרכזית ו כלי. | הוא מעורר פגיעה כרונית באיברי המערכת הלב וכלי הדם וחוסר יציבות במחזור הדם, מה שעלול להפוך לסיבות להופעת משבר יתר לחץ דם. | משפיע על גברים ונשים מעל גיל 40. |
| סוכרת |
הביטוי של סוכרת מלווה ב:
|
הפרעה מטבולית כרונית, המבוססת על ייצור לא מספיק של הורמון האינסולין, שאינו מסוגל להילחם בעלייה ברמות הגלוקוז בדם. | מוביל להפרעות מטבוליות, התפתחות תרדמת היפוגליקמית. | הוא מתפתח אצל נשים וגברים כאחד. |

מחלות אנוש תורשתיות הן פתולוגיות מולדות הנגרמות על ידי מוטציות גנטיות, שינויים במספר או הרס הכרום בשרשרת ה- DNA המאחסנת מידע גנטי, האחראי על ההתפתחות והמבנה הנכונים אדם.
בתורשה או כתוצאה מתקלה בגנים האבהיים או האימהיים, מחלות כאלה נחשבות לבלתי ניתנות לריפוי רובם קשורים ללקויות התפתחותיות חמורות, כמו גם רשימה גדולה של ליקויים חמורים במבנה החיצוני והפנימי של הגוף. אדם.
סרטונים על מחלות תורשתיות
מהן מחלות תורשתיות:


